
Pharmacogenomics and Precision Medicine Part 3
Steph’s Note: At long last, we are FINALLY ready to serve up the third and final installment in our pharmacogenomics and precision medicine series! If you haven’t pulled any pearls from Part 1 and Part 2 yet, you should definitely check those out to get your feet wet. Pharmacogenomics is a complex world with terminology that sends you reeling back to freshman year biology, wishing you could simply remember what the heck an allele is. OR…rather than driving yourself batty, you can just read the first 2 parts. Hint, hint.
Back with Part 3 is Dr. Saba Arshad. She’s lived a pretty diverse pharmacy life so far, which has led her to this crazy world of pharmacogenomics. Take it away, Saba, for the grand finale!
DPYD Pharmacogenomics for Fluorouracil and Capecitabine
Fluoropyrimidines like 5-fluorouracil (5-FU), capecitabine, and tegafur are widely used in cancer treatment. They target various cancers, such as colorectal, breast, and those of the aerodigestive tract. These drugs participate in complex metabolic pathways in the body. The genetic predisposition to severe toxicity from fluoropyrimidines stems largely from genetic variation in the DPYD gene, which encodes for the dihydropyrimidine dehydrogenase enzyme (DPD).
Let’s take a closer look at these pathways. Prepare yourself for some biochem chitty chat.
The activation of 5-FU involves conversion to fluorodeoxyuridine monophosphate (FdUMP), which inhibits thymidylate synthase (TYMS), a vital enzyme in DNA synthesis. This activation can occur via various pathways, involving enzymes like thymidylate phosphorylase (TYMP) and thymidine kinase.
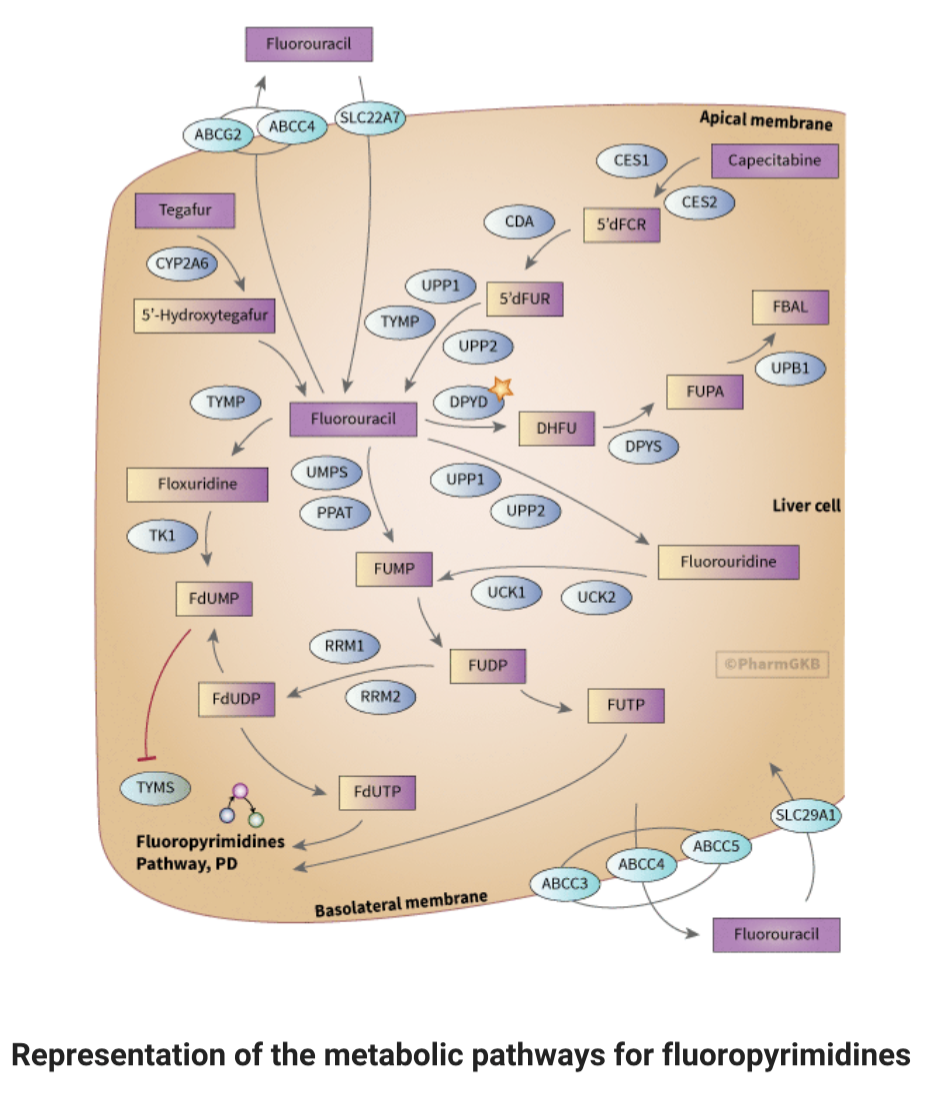
Yeah...it's THAT complicated. (Image)
5-FU is primarily metabolized in the liver, either directly when administered intravenously or indirectly after conversion from prodrugs like capecitabine and tegafur. Several enzymes play crucial roles in these metabolic pathways, with dihydropyrimidine dehydrogenase (DPD) being a key enzyme for converting 5-FU to its inactive form. Deficiencies in these enzymes can lead to increased systemic drug exposure and severe toxicity.
However, tumors can develop resistance to these drugs, partly due to changes in gene expression such as TYMS and P53. Additionally, alterations in drug transporters, like SLC29A1, ABCG2, ABCC3, ABCC4, and ABCC5, can also contribute to drug resistance.
Geez. Fluoropyrimidines don’t mess around.
They require a LOT of steps to take action in the body AND even more steps to get out of it. Check out the metabolic pathways of fluoropyrimidines in the image to the right here. Sure, we’re not going to test you on every detail in this figure (you’re welcome!), but having at least a general understanding of the complex pharmacokinetics and pharmacodynamics of fluoropyrimidines is crucial for optimizing their efficacy and minimizing toxicity in cancer treatment.
Gene #1: DPYD and Fluoropyrimidine Pharmacogenomics
DPD enzyme deficiency is a condition related to a deficit in the metabolism of thymine and uracil, resulting in excessive amounts of uracil and thymine in the blood, urine, and CSF. It can be inherited as an autosomal recessive trait, such as homozygosity for deleterious DPYD variants. Patients with homozygous DPYD variants can manifest with severe DPD deficiency. (Remember, DPD is the enzyme encoded by the DPYD gene.)
During infancy, this can result in neurologic and other severe defects. It is a rare condition that has been documented mostly in single case reports. However, some people are also asymptomatic.
The prevalence of DPD deficiency, based on activity levels measured in blood cells and the uracil breath test, has been reported as 3%-5% in patients of European origin and 8% in patients of African origin. In addition, the US National Library of Medicine reports a prevalence of partial deficiency of 2%-8%, based on the aforementioned study and others. So what’s the clinical impact of these numbers?

Snoop can't believe those numbers either. (Image)
The US National Institutes of Health has estimated that approximately 1,300 deaths per year are attributable to DPD deficiency, on the basis of a prevalence of 0.5% deaths in patients treated with fluorouracil annually.
Wow. That’s a lot for something many of us don’t know about…which is why I’m here to raise a flag! Ok, so shouldn’t we be testing or something? There’s gotta be something we can do to help prevent 1,300 deaths every year!
Thanks for asking! Yes, we can test for DPYD variants. The specificity and sensitivity of DPYD genetic testing is as follows:
Testing for p.D949V has a sensitivity of 2%, specificity of 100%, positive predictive value of 86%, and negative predictive value of 51%.
Testing for *2A, *13, and p.D949V combined has similar rates of predictive values. This is because patients who are carriers of these alleles have a high risk of developing severe toxicity (high specificity).
How do we use these results to guide our clinical decision making? Glad you asked. Check out the below chart of possible DPYD variants and associated recommendations:

(Image)
There’s also a somewhat simplified system for assigning a patient a DPYD activity score, which can help to determine how much fluoropyrimidine should be administered:
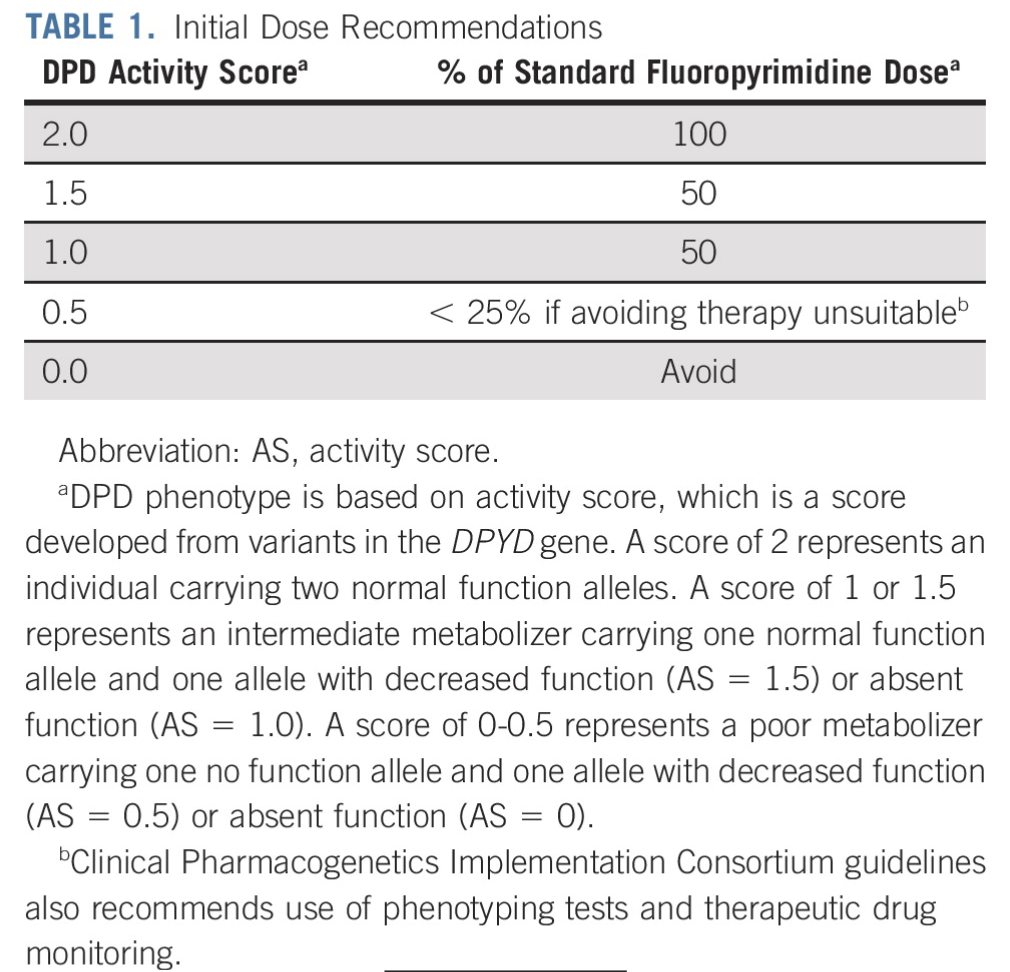
(Image)
DPYD Testing and Oncology Guideline Recommendations
In the National Comprehensive Cancer Network (NCCN) guidelines, the risk of severe toxicity conferred by DPYD variants is acknowledged, but pretreatment genotyping for DPYD variants is not supported because the evidence behind it is deemed to be controversial. Specifically, the NCCN colon cancer guideline acknowledges that pretreatment DPYD testing can identify the 1%-2% of the population with specific alleles indicating a higher risk of severe toxicity. However, this guideline is too narrow, as other DPYD variants also justify testing, potentially raising the number of at-risk patients to about 9% of the US population.
The American Society of Clinical Oncology (ASCO) also does not provide any guidelines on DPYD testing.
The US Food and Drug Administration (FDA) package inserts for capecitabine and fluorouracil (FU) acknowledge patients with DPD deficiency have an increased risk of life-threatening toxicity; however, instead of recommending preemptive testing, it is recommended for people who have a known DPD deficiency to discuss it with their physicians. In March 2024, FDA finally updated the labeling changes in regards to fluorouracil products, but there are still no specific pharmacogenomic testing recommendations included in the verbiage.
Thankfully, on April 30, 2020, the European Society for Clinical Oncology “recommended that patients should be tested for the lack of the enzyme DPD before starting cancer treatment with fluorouracil given by injection or infusion (drip) or with the related medicines, capecitabine, and tegafur.” The French National Agency for the Safety of Medicines and Health Products and the Medicines and Healthcare Products Regulatory Agency also have each approved guidelines for preemptive DPYD testing for patients treated with fluoropyrimidines.
In case you need more convincing, check out a couple of the below vignettes. Remember, this is preventable.
In 2018, a patient received capecitabine without prior testing for dihydropyrimidine dehydrogenase (DPD) deficiency and later presented with vomiting, rash, and diarrhea. The hospital failed to provide uridine triacetate (the rescue medication) in a timely fashion, and the patient died. The patient’s widow filed a wrongful death lawsuit against Oregon Health Sciences University (OHSU) and assisted in the formation of a nonprofit organization to advocate for DPYD testing for fluoropyrimidines. A settlement for $1 million was reached, which also requires OHSU oncologists to undergo education about DPYD testing and inform their patients about its availability.
Here’s yet another heartbreaking incident. This happens. It’s not just a textbook pearl. It’s real.
So what’s the hold up on including pre-emptive genetic testing in American guidelines?
Like all genetic tests, levels of evidence vary for each allele. NCCN’s primary objection to testing DPD activity involves uncertainty that every patient with low DPD activity is at risk and the degree to which DPYD variants confer such risk. In public commentary, NCCN board members state further studies are required to mitigate the possibility that dose reduction would reduce fluoropyrimidine efficacy in some patients.

(Image)
However, I would counter that current evidence suggests dose adjustments do not alter efficacy; thus, a requirement for additional efficacy research should not supersede established concerns of unacceptable rates of life-threatening toxicity in DPYD low-activity variant carriers.
Sufficient data are now published in the literature to conclude that individuals harboring certain DPYD variants are at increased risk of toxicity or death when administered standard doses of fluoropyrimidines. A recent 2021 meta-analysis of 13,929 patients in 35 studies found that patients carrying DPYD*2A were much more likely to experience severe life-threatening toxicity from fluoropyrimidine therapy than those carrying only wild-type alleles.
Although this continues to be a controversial pharmacogenomics issue, hopefully you now have a better understanding of the clinical scenario and can be an advocate for your fluoropyrimidine patients!
Now on to pain management…
Opioids and Pharmacogenomics
Opioids are most commonly used to control acute and chronic pain. The therapeutic effect and adverse event profiles vary largely due to interindividual variability. According to the US Department of Health & Human Services, in 2019, 22.1% of U.S. adults with chronic pain had used a prescription opioid in the previous 3 months. Uninsured adults had a lower prevalence of prescription opioid use (12.1%) than adults with private coverage (19.9%), Medicare (28.2%), or Medicaid or other forms of public coverage (28.4%).
So how do genetics play a role in opioid use and optimization?
Gene #1: CYP2D6 Polymorphisms and Opioid Pharmacogenomics
Several common opioids—specifically, codeine, hydrocodone, methadone, oxycodone, and tramadol—are metabolized by the CYP2D6 enzyme.
CYP2D6 converts codeine and tramadol to their more active metabolites, morphine and O-desmethyltramadol, respectively. Therefore, alterations in the metabolism of these drugs can have a profound impact on their analgesic effects or risk of side effects.
Oxycodone and hydrocodone are also converted by CYP2D6 into oxymorphone and hydromorphone, respectively. Although oxymorphone and hydromorphone have higher morphine equivalents than their parent drugs, both oxycodone and hydrocodone are active drugs. So alterations in their metabolic pathways have a less profound clinical effect than observed with codeine and tramadol.
Methadone, although partially metabolized by CYP2D6, is predominantly metabolized by other enzymes.
For you visual folks:
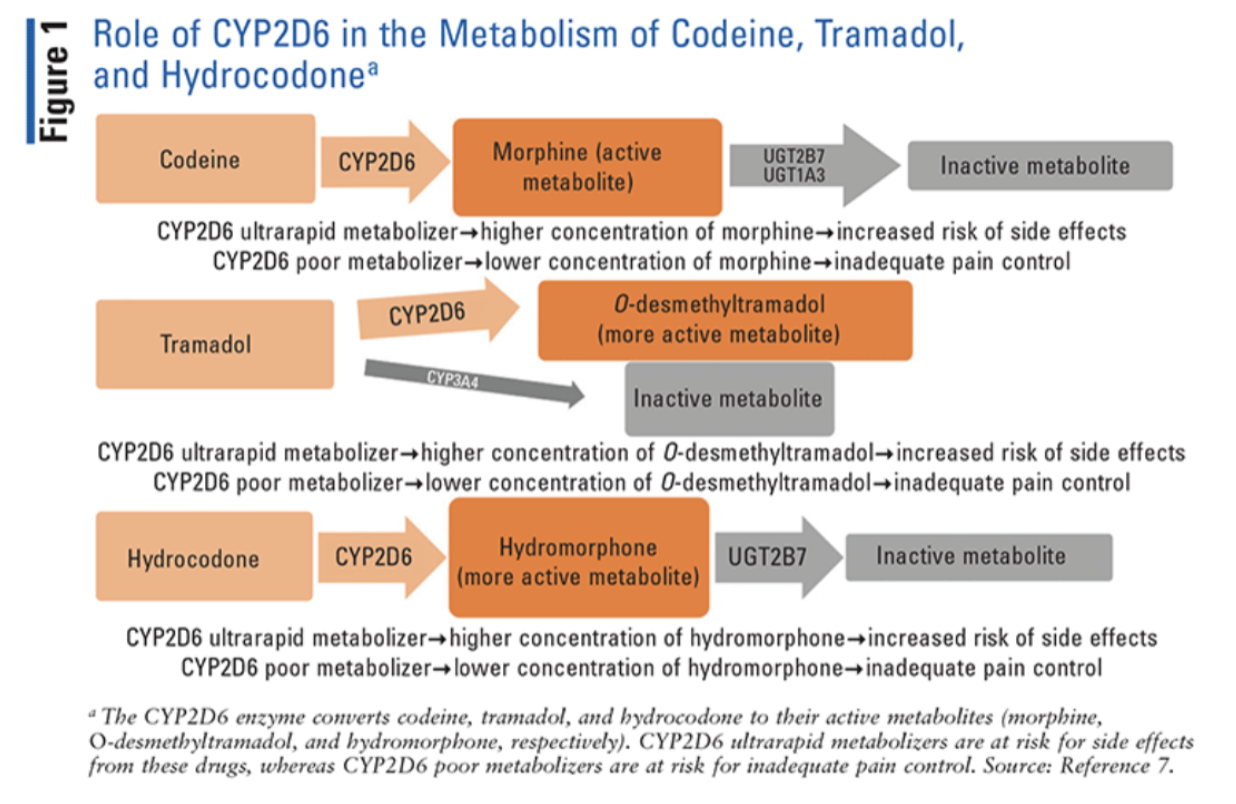
(Image)
CYP2D6 is a highly polymorphic gene and has over 130 alleles. The most commonly reported alleles are categorized into functional groups as follows:
Normal function (e.g., CYP2D6*1, *2, and *35)
Decreased function (e.g., CYP2D6*9, *10, *17, *29, and *41)
No function (e.g., CYP2D6*3–*6)
The CYP2D6*5 allele represents a deletion of the gene from one allele, resulting in a no function allele. FYI, gene duplications and multiplications are denoted by “xN” (e.g., CYP2D6*1xN with xN representing the number of CYP2D6 gene copies in cis).
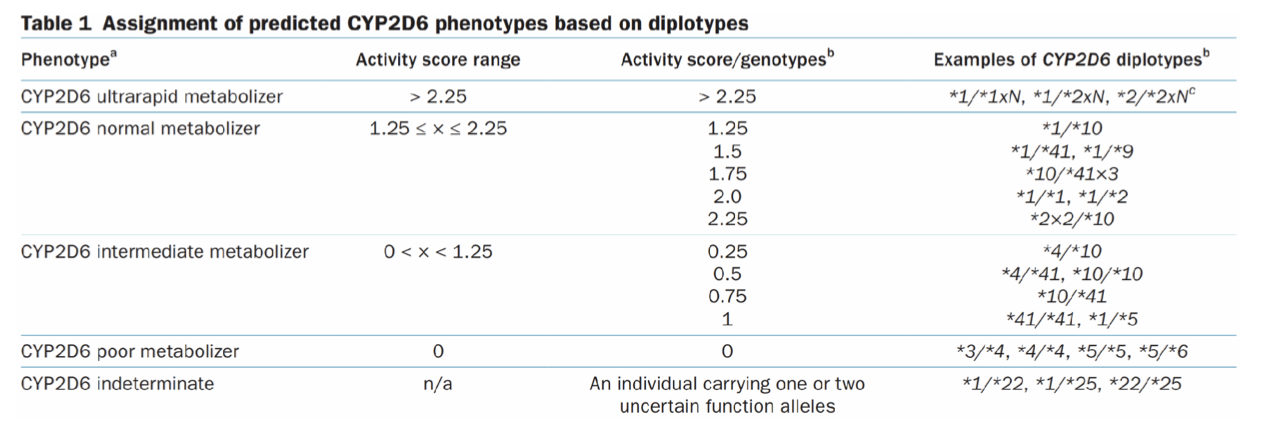
For those of you who crave the deets, here's all the info of CYP2D6 phenotypes. (Image)
Pharmacogenomics-guided opioid therapy enables clinicians to determine the best options for avoiding treatment failure and serious side effects. However, clinicians should continue to personalize pain management by considering other factors as well. For example, elderly patients are at higher risk for falls and fractures secondary to oversedation. Therefore, opioid therapy in elderly patients should be initiated and titrated with caution, and patients should be monitored more frequently for constipation, cognitive dysfunction, sedation, and fall risk. Check out the table below for additional special considerations:
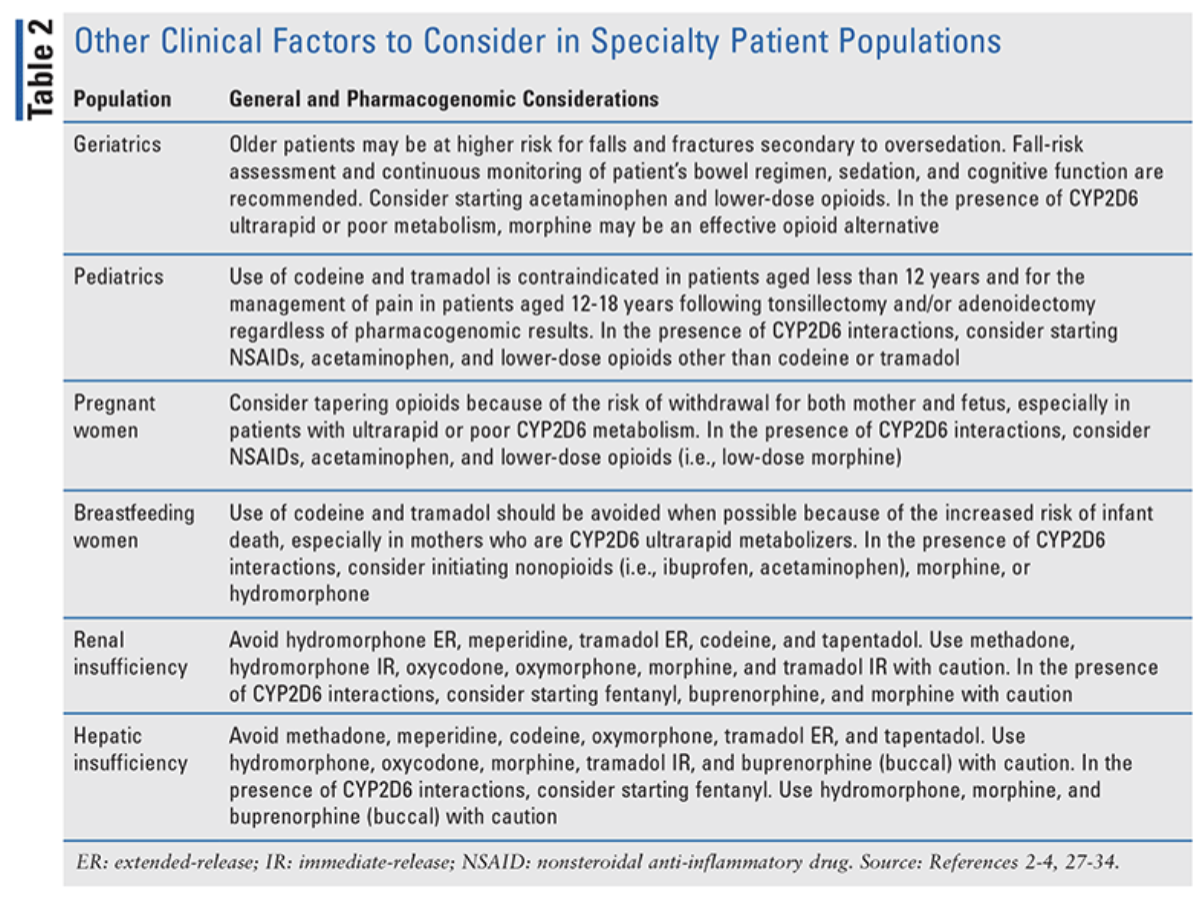
(Image)
Risks of oversedation and respiratory depression are considerable if opioid therapy is used in conjunction with additional CNS depressants, such as muscle relaxants and benzodiazepines. Particular caution should be taken when CYP2D6 substrates are used in the presence of significant drug-drug interactions, especially CYP2D6 inhibitors (e.g., bupropion, cinacalcet, fluoxetine, paroxetine, quinidine) and inducers (e.g., dexamethasone, oritavancin, rifampin). This is because interacting medications can potentially change the CYP2D6 phenotype (i.e., a phenomenon known as phenoconversion).
An alternative opioid with metabolism independent of CYP2D6 may be safer in patients with interacting medications.
A Note on Phenoconversion
Phenoconversion is a phenomenon that converts genotypic extensive metabolizers (EMs) into phenotypic poor metabolizers (PMs) of drugs, thereby modifying their clinical response. An ironic aspect of drug-induced phenoconversion is that a relatively high number of patients require concurrent medications that result in phenoconversion!
One notable example that would help to understand this process…

See the eagle...beeee the eagle... Pharmacist eagle eyes to the rescue! Spot those drug interactions! (Image)
Strong (e.g., paroxetine, bupropion) and moderate (e.g., duloxetine, sertraline) inhibitors can also decrease the CYP2D6 activity, phenoconverting the genotypic normal metabolizers (NMs) or ultra-metabolizers (UMs) into phenotypic PMs or intermediate metabolizers (IMs). So, concomitant use of those drugs cannot be overlooked while predicting the CYP2D6 phenotype. Moreover, data indicate that up to 20–70% of patients who are on CYP2D6 metabolized drugs are at risk of CYP2D6 phenoconversion by concomitant medications.
Considering the importance of phenoconversion, Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines suggest incorporating the effects of CYP2D6 inhibitors while calculating the activity score of CYP2D6 for CYP2D6 metabolized opioids, tricyclic antidepressants, tamoxifen, and atomoxetine.
Genes #2 & 3: COMT and OPRM1 Polymorphisms and Opioid Pharmacogenomics
The recent CPIC opioid guideline highlights potential associations of the OPRM1 and COMT genes with altered opioid efficacy and adverse effects (AEs). Alterations in OPRM1, which codes for mu receptors, can reduce their expression, while COMT affects pain perception by methylating catecholamines like dopamine. Polymorphisms in COMT can lead to higher pain levels and reduced opioid effectiveness.
Although some studies indicate that patients with certain OPRM1 polymorphisms might need higher opioid doses, there is no evidence linking COMT variants to increased opioid AEs. Due to inconsistent clinical findings, CPIC has not recommended opioid dosing adjustments based on these genetic polymorphisms, although this may change as more evidence emerges.
Challenges to Implementation of Pharmacogenomic Testing for Opioid Use
Proper interpretation of genotype-based phenotype from CYP2D6 alleles or activity score.
Adjustment of phenotype considering concomitant administration of CYP2D6 inhibiting medications.
Routine checking of concomitant medications for phenotype adjustment.
Reimbursement of genetic testing costs.
Proper training of clinicians.
Abacavir and HLA-B Pharmacogenomics
Abacavir is a nucleoside reverse transcriptase inhibitor indicated for the treatment of HIV infection, in combination with other medications, as part of highly active antiretroviral therapy. Abacavir competitively inhibits viral reverse transcriptase, suppressing HIV’s ability to convert its RNA genome into DNA before insertion into a host cell’s genome.
It is commercially available as a single agent (Ziagen) or co-formulated as a fixed dose combination with other nucleoside reverse transcriptase inhibitors:
with lamivudine (Epzicom),
with lamivudine and dolutegravir (Triumeq), and
with lamivudine and zidovudine (Trizivir).
For more details on HIV treatments, including abaavir, check out this handy post.
Gene #1: HLA-B*57:01 and Abacavir Hypersensitivity
Human Leukocyte Antigen B (HLA-B) is responsible for presenting peptides to immune cells and plays a critical role in normal immune recognition of pathogens. HLA-B is part of the major histocompatibility complex (MHC) gene family located on chromosome 6.
HLA genes are highly polymorphic, and more than 1,500 HLA-B alleles have been identified. A variant allele, HLA-B*57:01, is associated with increased risk of a hypersensitivity reaction to the anti-HIV drug abacavir. In the absence of genetic prescreening, hypersensitivity affects ~6% of patients and can be life-threatening with repeated dosing.

(Image)
The frequency of the HLA-B*57:01 allele is lowest in African and Asian populations. It is totally absent in some African populations, as well as in the Japanese. In European populations, this allele is relatively common, with a frequency of 6–7%. The highest frequency of HLA-B*57:01 is reported in Southwest Asian populations, where up to 20% of the population are carriers.
Abacavir hypersensitivity reactions can vary in presentation. But here’s the general overview of possibilities. If a patient has at least 1 symptom from 2 or more of the following groups, they are supposed to seek advice ASAP from their health care provider:
Group 1: Fever
Group 2: Rash
Group 3: Nausea, vomiting, diarrhea, abdominal pain
Group 4: Generally ill feeling, extreme tiredness, or achiness
Group 5: Shortness of breath, cough, sore throat
Sounds kind of like your general, non-specific, flu-like illness, right? Or maybe even COVID? Tricky tricky… But that’s why patients on abacavir should always be counseled on possible signs and symptoms of hypersensitivity as well as knowing that it’s always better to call than not if they’re unsure.
Recommendations for Use of Pharmacogenomics with Abacavir
The results of the 2008 PREDICT-1 study, which was the first double-blind, prospective, randomized trial of a genetic test aimed at reducing adverse drug events, revealed that genetic prescreening for HLA-B*57:01 led to no immunologically confirmed hypersensitivity reactions (HSR) among patients who tested negative for HLA-B*57:01 in the genetic testing arm. This was compared to a 2.7% incidence of immunologically confirmed HSR among 842 unscreened patients in the standard-of-care control arm.

The tl;dr of HLA-B genotype and use of abacavir. (Image)
These findings prompted the FDA to implement a black box warning in 2008 regarding the high risk of HLA-B*57:01-associated HSR. The FDA recommended that all patients undergo screening before being treated with abacavir, even those who had previously tolerated the drug and were being restarted on therapy. Additionally, the FDA advised against initiating abacavir treatment in carriers of HLA-B*57:01.
Abacavir stands out as one of the few drugs for which the FDA has recommended genetic testing prior to use, serving as a notable example of pharmacogenetics being integrated into routine medical practice. Yayy!! :)
Of note, abacavir skin patch testing may be performed after a case of clinically diagnosed HSR to determine whether it can be immunologically confirmed. At this time, skin patch testing is an investigational procedure, and the results should be interpreted only by an experienced immunologist.
And there you have it! That’s the end of Part 3 of our Pharmacogenomics and Precision Medicine series. I hope this has introduced you to the fascinating world of genetics and medicine, as well as giving you the tools and resources to learn more should you need or want to. Happy genetic screening!