
Pharmacogenomics and Precision Medicine Part 2
Steph’s Note: This week, we’re returning to a somewhat lonely space on tl;dr…pharmacogenomics. Outside of our initial post on this topic last year, we haven’t really done much (erm…anything…). Which, let me be clear, reflects our general lack of expertise on the subject rather than lack of importance! But thankfully, Dr. Saba Arshad has deemed tl;dr worthy enough of her time to come back for round 2, which includes updates from Part 1 as well as brand, spanking new info on everything from warfarin to statins to anesthetics. Take it away, Saba!
Warfarin and Pharmacogenomics
Sure, we live in the age of DOACs, but we can’t totally leave our old frenemy warfarin completely out of the picture! Even though pharmacy students likely don’t have multiple case studies on dosing warfarin anymore (although they should…), it still has its place for many patients. (If you need a refresher or - no judgment - an introduction, check out this post.) So let’s talk about one major piece of the warfarin puzzle.
Warfarin is a widely used anticoagulant with a narrow therapeutic index and large interpatient variability when it comes to the dose required to achieve target anticoagulation. Common genetic variants in CYP2C9, VKORC1, CYP4F2, and the CYP2C cluster (e.g., rs12777823), as well as multiple non-genetic factors, account for 50% of warfarin dose variability.

And you thought relationships were complicated… try warfarin dosing. (Image)
So how is one supposed to ever get warfarin dosing right?
Warfarin dosing typically starts with an empirical initial dose, followed by at least weekly INR measurements and dose adjustments. The initial dose often averages around 4–5 mg/day, but sometimes loading doses are used initially (hello, orthopedic and vascular surgeons). Stable daily doses to achieve an INR of 2–3 can range from 1–20 mg.
Yes, you read that correctly. Some patients take up to 20 mg of warfarin a day! Wowsers.
In order to talk pharmacogenomics of warfarin, you gotta at least know the basics of how warfarin works:

Warfarin is administered as a racemic mixture, which inhibits vitamin K epoxide reductase complex. S-warfarin is about 5 times more potent than R-warfarin at doing this job. Affecting the metabolism of the enantiomers of warfarin, especially S-warfarin, significantly impacts the anticoagulation induced by the drug. The genes with the strongest literature support (and for which we actually have recommendations for use in warfarin dosing) are…
CYP2C9,
VKORC1, and
CYP4F2.
Additionally, genomewide association studies have identified an independently significant single nucleotide polymorphism (SNP) in the CYP2C cluster, which we will briefly discuss as well. But starting with CYP2C9…
Gene #1: CYP2C9 and Warfarin Pharmacogenomics
CYP2C9 is a hepatic drug-metabolizing enzyme in the cytochrome P450 (CYP450) superfamily, which is the primary metabolizer of S-warfarin. The two most common decreased function alleles among individuals of European ancestry are:
CYP2C9*2 (c.430C>T; p.Arg144Cys; rs1799853)
CYP2C9*3 (c.1075A>C; p.Ile359Leu; rs1057910)
Ok, before we get too far, we may need to circle the wagons on what all the asterisks are for, especially if you’re new to genetic testing. Here’s a quick rundown on a few terminology essentials for this post:
Star alleles: Versions of the same gene that have different DNA spelling changes compared to each other.
Diplotypes: A specific combination of two haplotypes (see haplotype definition below).
Haplotype: A combination of multiple spelling changes within a particular gene.
CYP2C9 allele frequencies differ between racial/ethnic groups. In vitro and in vivo studies suggest CYP2C9*2 and *3 impair metabolism of S-warfarin by 30–40% and 80–90%, respectively. Take a moment for that latter number to sink in… that means metabolism of the potent anticoagulant enantiomer can be impaired up to 90%!
Compared to patients homozygous for CYP2C9*1, individuals who inherit one or two copies of CYP2C9*2 or *3 are at greater risk of bleeding during warfarin therapy, require lower doses to achieve similar levels of anticoagulation, and need more time to achieve a stable INR. Aka, they have VERY clinically significant differences in their warfarin needs than the average bear! These alleles are found with the highest frequency among those of African ancestry.
Gene #2: VKORC1 and Warfarin Pharmacogenomics
VKORC1 encodes the vitamin K epoxide reductase protein, the target enzyme of warfarin. VKORC1 catalyzes the conversion of vitamin K-epoxide to vitamin K, which is the rate-limiting step in vitamin K recycling. A common variant upstream of VKORC1 (c.-1639G>A, rs9923231) is significantly associated with warfarin sensitivity.
Patients with one or two–1639A require progressively lower warfarin doses than–1639G/G homozygotes. The–1639G>A polymorphism is present on a haplotype that affects VKORC1 protein expression.
Gene #3: CYP4F2 and Warfarin Pharmacogenomics
Well, this is a new one to you, eh?! While it’s not uncommon to be somewhat familiar with the effects of genetic variations in CYP2C9 and VKORC1 on warfarin dosing, CYP4F2 doesn’t usually hit the radar. But it’s actually a really important part of the warfarin story!
CYP4F2 is a primary liver vitamin K oxidase that catalyzes the metabolism of vitamin K to hydroxy-vitamin K1 and removes vitamin K from the vitamin K cycle. It acts as an important counterpart to VKORC1 in limiting excessive accumulation of vitamin K.

Sing it with me. “Make waaay for Prince CYP4F2!” (Image)
Importantly, including the CYP4F2 variant in warfarin dosing models that already incorporated CYP2C9, VKORC1, and clinical factors actually improved the accuracy of dose prediction. This correlation has been confirmed in subsequent studies with those of European and Asian ancestry, although not those of African ancestry.
So, BAM. Move over CYP2C9 and VKORC1. Make way for CYP4F2.
A SNP to Know for Warfarin Pharmacogenomics
Remember how I said earlier that in addition to the genetic variations in the 3 enzymes there is a SNP that also affects warfarin dosing? Well, here it is.
CYP2C rs12777823 is a SNP in the CYP2C cluster near the CYP2C18 gene on chromosome 10 and is associated with a clinically relevant effect on warfarin dose through significant alterations in warfarin clearance, independent of CYP2C9*2 and *3.
Research shows that a specific SNP influences warfarin dosage in African Americans but not in other ethnic groups or Egyptians. This suggests the SNP may be linked with other variants affecting warfarin response uniquely in African Americans, who are primarily of West African descent. However, it’s unclear if this association exists in other African populations.
So many pieces to the warfarin puzzle… and now that’s just one more to know.
Pulling Together Warfarin Pharmacogenomics into Actionable Recommendations
So what on earth is a pharmacist to do with all these genetic and non-genetic warfarin dosing influencers floating around? How can we possibly turn these jumbled letters and numbers into actual therapeutic recommendations?
Luckily, great minds have already done this for us. Check out this Pharmacogenetic Warfarin Dosing Algorithm:

Pharmacogenetic warfarin dosing should use specific algorithms, and the above figure provides dosing recommendations based on genotype for adults. A few notes…
“Dose clinically” means dosing without genetic info, possibly using a clinical algorithm or standard dose.
Data is most robust for European and East Asian ancestry and consistent in other populations.
45-50% of individuals with African ancestry have certain CYP2C9 variants; if these aren’t tested, dose warfarin clinically.
Most algorithms target an INR of 2-3.
Consider alternative agents for genotypes indicating poor metabolism or increased sensitivity to warfarin.
The EU-PACT trial offers a loading dose algorithm, not yet validated for African ancestry.
Prescriptions adjust doses to practical regimens (e.g., 4.3 mg/day could be 4 mg daily and 5 mg twice weekly). An additional dose revision algorithm, usable on days 4-5, incorporates genetic info and has been tested in COAG and EU-PACT trials.
The warfarindosing.org website contains the Gage algorithm as primary and IWPC as secondary, both adjustable for certain genotypes.
Some additional recommendations and limitations of warfarin genetic dosing algorithms:
Non-African Ancestry Recommendations:
Use pharmacogenetic algorithms to calculate doses, incorporating VKORC1 and CYP2C9 genotypes.
Consider alternative anticoagulants for those with poor metabolism or increased sensitivity.
Genetic variants like CYP2C9*5, *6, *8, or *11 may also require dose adjustments.
Increase doses by 5-10% if the CYP4F2*3 alleles are detected.
African Ancestry Recommendations:
For those with variants CYP2C9*5, *6, *8, or *11, reduce doses by 15-30%.
Include rs12777823 genotype information, recommending dose reductions of 10-25% for certain genotypes.
Pediatric Recommendations:
Strong evidence supports using VKORC1 and CYP2C9 genotypes for dosing in European ancestry children.
Data for other ethnicities and additional genetic variants are limited.
Now remember (you know there’s always a disclaimer, right??)… Genetic information can improve dosing accuracy and reduce adverse events, but it should NOT replace regular INR monitoring. Additionally, the benefits of genetic testing and using the algorithm are most significant at therapy initiation. Long-term, stable patients may see limited benefit from genetic testing because why try to fix something that isn’t broken?
Also, patients with variable adherence may see little benefit from genetic testing (because the problem isn’t necessarily getting the right dose…but TAKING the right dose….) Don’t forget that these tests cost money, money, money! On that note, genetic testing can be cost-effective but is not universally covered by insurance.
To add insult to the risk of injury with pharmacogenomics, there’s also the potential for false security in genetic dosing and risk of misdosing if rare variants are not considered. Basically, you still have to have your clinical judgment hat on and not trust any test 100%. Errors in genotyping can have long-term health impacts.
So there you have it. The story of warfarin and genetics, in a GIANT nutshell. Hopefully this adds to your anticoagulation arsenal. Now on to our next class of medications in the story of pharmacogenomics: statins.
Statins and Pharmacogenomics
Statins are crucial medications that reduce cholesterol and prevent cardiovascular diseases, making them some of the most commonly prescribed drugs globally. Imagine statins as meticulous housekeepers, ensuring your heart stays spotless and clutter-free!
One challenge associated with statin therapy is statin-associated musculoskeletal symptoms (SAMS). This term encompasses muscle aches, cramping, pain, weakness, and even creatinine kinase (CK) elevations. SAMS can affect adherence, potentially reducing the long-term effectiveness of statin therapy.
So what role does genetics have in SAMS?
There are several genetic factors that may influence statin response, including:
SLCO1B1: This gene encodes a transporter that facilitates hepatic uptake of all statins
ABCG2: Encodes an efflux transporter affecting the absorption and disposition of rosuvastatin
CYP2C9: Encodes for a phase 1 drug metabolizing enzyme that is responsible for oxidation of statins
Genetic variations in these genes can alter the body’s exposure to different statins (simvastatin, rosuvastatin, pravastatin, pitavastatin, atorvastatin, fluvastatin, and lovastatin), potentially increasing the risk for SAMS. Let’s look at each of these in a little more detail.
Gene #1: SLCO1B1 and Statin Pharmacogenomics
As previously stated, SLCO1B1 encodes a transporter that aids in hepatic uptake of statins. Alterations in this gene and encoded protein can lead to increased exposure to statins, which can increase risk for SAMS. Genetic variability in SLCO1B1 also influences the hepatic uptake of other drugs (e.g., methotrexate) as well as important endogenous compounds (e.g., bilirubin). Complete SLCO1B1 and SLCO1B3 deficiency is associated with Rotor syndrome, a type of hyperbilirubinemia.
Check out the below summarized version of assignment of the predicted SLCO1B1 phenotype based on star (*) allele diplotypes:

(Image)
How has this information been translated into actionable recommendations?
The American College of Cardiology and the American Heart Association issued an updated clinical practice guideline for the management of blood cholesterol in 2018. In the guidelines, it is recommended to initiate high-intensity statins in patients with evidence of clinical atherosclerotic cardiovascular disease, which may include atorvastatin 40 or 80 mg once daily or rosuvastatin 20 or 40 mg once daily. But what about patients with SLCO1B1 variations?

The figure on the left is for SLCO1B1 DECREASED function phenotypes, whereas the figure on the right is for SLCO1B1 POOR function phenotypes. Statins and statin doses indicated in the light grey boxes can be prescribed with the lowest risk for SAMS. (Image)
So that’s the SLCO1B1 story. On to the next gene!
Gene #2: ABCG2 and Statin Pharmacogenomics
ABCG2 encodes an efflux transporter, which specifically affects rosuvastatin and atorvastatin out of all of the statins. Genetic polymorphisms in ABCG2 also influence absorption and disposition of many other non-statin drugs, including anticancer drugs and antiviral drugs. In addition, genome-wide association studies reveal that ABCG2 variants influence serum uric acid levels, risk for gout, and response to the antigout medication, allopurinol.
Check out this summary of the ABCG2 phenotypes:

FYI Unlike SLCO1B1 and CYP2C9, there is no star allele nomenclature to represent ABCG2 variants at this time. (Image)
What are the actionable recommendations for us pharmacists? How are we to put this information into practice for our patients?
You’re in luck - we have specific guidance! But FYI, these recommendations for ABCG2 variants are specific to rosuvastatin. Although atorvastatin pharmacokinetics are also affected by ABCG2 genetic variation, at this time there is insufficient evidence to provide therapeutic recommendations.

(Image)
A few key points:
Likely because of the higher hepatic exposure, the ABCG2 c.421AA variant has also been associated with improved cholesterol lowering response to rosuvastatin in large genome-wide association studies.
Selection and dosing of rosuvastatin should also consider Asian ancestry.
Aaaaand, now on to the third gene to know for statins!
Gene #3: CYP2C9 and Statin Pharmacogenomics
CYP2C9 encodes an enzyme responsible for oxidation of statins (part of their metabolism). Let’s take a quick look at the various phenotypes of the CYP2C9 variations, as pertains to statins:
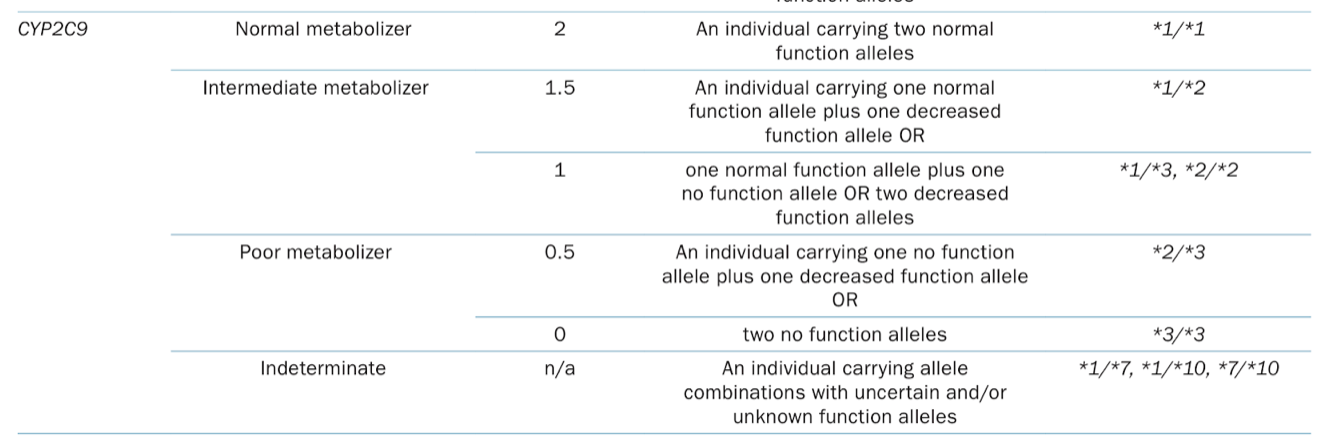
Back to the * allele nomenclature for CYP2C9, just like we saw when we discussed this enzyme and warfarin earlier. (Image)
At this point, statin dosing recommendations for CYP2C9 variants are specific to fluvastatin. (Other statins’ pharmacokinetics and pharmacodynamics are not thought to be influenced by CYP2C9.) Here are those dosing recs:

Additional Considerations when Using Genetic Tests for Statin Therapy
Although clinicians are accustomed to trials of discontinuing and reinitiating statin therapy in those who develop SAMS, in many patients statin therapy is never resumed. As a result, LDL cholesterol values are higher, as is their risk for cardiovascular disease. Careful weighing of risk versus benefit is imperative for each individual patient to ensure they’re not missing out on beneficial therapy.
Additionally, this is where pre-emptive genetic testing could be hugely helpful! Pre-emptive SLCO1B1, ABCG2, and CYP2C9 testing may lead to a reduction in the incidence of SAMS, by identifying those at significant risk and recommending a lower statin dose or an alternative statin with lower SAMS risk. This could mean more people stay on their beneficial statin therapy by sidestepping the SAMS issue altogether.
While prospective data showing that prescribing based on genetic testing results alters SAMS incidence are lacking, there are emerging data demonstrating an improvement in patients’ perceptions of statins, appropriate statin prescribing, neutral data on patient-reported adherence, and mixed data on reducing LDL- cholesterol levels.
That being said, as with any diagnostic test, genetic variation is just one factor that clinicians should consider when prescribing statins. Furthermore, rare variants may not be included in the genotype test used, and patients with rare variants that reduce SLCO1B1 function may be incorrectly assigned a normal phenotype based on a default to wild-type (*1) test result. However, this risk can be minimized by…
Monitoring to ensure that the appropriate LDL cholesterol reduction is achieved for the intended statin intensity, and
Using an alternative statin with a similar statin intensity based on the recommendations.
Another potential risk to statin genetic testing is that a patient or provider may inappropriately stop or avoid statin therapy based on the results, and this could cause higher LDL cholesterol and increased cardiovascular risk.
That would be…unfortunate…
Alright, now that you are experts in warfarin and statin pharmacogenomics, we’re going to move on to the last section of Part 2 - anesthetics.
Pharmacogenomics and Malignant Hyperthermia
Potent volatile anesthetic agents are widely used and generally safe agents for inducing general anesthesia. These anesthetics include the following:
Sevoflurane
Halothane
Enflurane
Isoflurane
Methoxyflurane
Desflurane
All of the currently available potent inhalation anesthetics are presumed to be equivalent triggers of malignant hyperthermia (MH). For funsies, here’s a little more about the history of inhaled anesthetics:

(Image)
Of note, MH may also be triggered by depolarizing neuromuscular blockers like succinylcholine. This agent binds to acetylcholine receptors at the neuromuscular junction, which triggers channel opening, causing initial muscle activation followed by sustained depolarization. This process results in profound muscle relaxation, which is beneficial for procedures like intubation and surgery.
If you’re like most of us, you probably didn’t learn much about anesthetics’ mechanisms in pharmacy school, so let’s take a quick look at how they work.
Mechanism of Action of Inhaled Anesthetics
In short (because this is tl;dr after all), inhaled anesthetics are thought to make it more difficult for neurons to depolarize and propagate signals from the spinal cord to the cerebral cortex (aka, inhibition of afferent signals). They also reduce motor response to pain (aka, inhibition of efferent signals). They induce hypnosis and amnesia, as well as decrease CNS blood flow and glucose metabolism.
All of these effects are mediated (allegedly…because it’s not fully elucidated) by enhancing inhibitory pathways and depressing excitatory pathways. For the former, inhaled anesthetics enhance GABA(a) receptor activity, leading to increased chloride transport and hyperpolarization of the neuronal cell membrane. This means it’s harder for those cells to depolarize and send signals. These agents are also thought to affect a specific type of potassium channel to the same end effect: hyperpolarization and increased depolarization threshold.
For the latter mechanism, these agents are thought to block ion transport through excitatory NMDA (aka, glutamate), nicotinic, and serotonergic receptors to reduce neuronal signal propagation. Basically, road blocks and traffic jams to slow it all dowwwwwwn.
Alright, now that we have the basics of how these agents work, let’s circle back around to the issue with them: malignant hyperthermia. What is that??
What is Malignant Hyperthermia Susceptibility (MHS)?
MH is estimated to occur in 1:100,000 surgical procedures in adults and in 1:30,000 pediatric procedures. The exact mechanism by which MHS pathogenic genetic variants cause MH is not known. But current evidence strongly suggests that upon exposure of an MH-susceptible person to a triggering agent, there can be an increased influx of cytoplasmic calcium within skeletal muscle fibers, which leads to uncontrolled muscle contractions. Interestingly, about 1:2000 people have one of these predisposing genetic variants!
The most sensitive and early indicators of MH are:
tachycardia and an increase in end-tidal CO2 followed by skeletal muscle rigidity,
metabolic and respiratory acidosis,
hyperkalemia, and
hyperthermia and arrhythmia.
If left untreated, an MH reaction can result in cardiac arrest and death.

MH patient in the process of being cooled down. (Image)
What’s the treatment? Dantrolene, dantrolene, dantrolene! This medication attempts to reverse the uncontrolled muscle contractions of MH by blocking calcium release from the sarcoplasmic reticulum, which helps to promote muscle relaxation. Other treatments include supportive oxygen and cooling in the form of cold IV fluids and/or ice packs.
So what are the pathogenic genetic variants?
Gene #1: RYR1 and Malignant Hyperthermia Susceptibility
The RYR1 protein is a key component of the calcium release channel found in the sarcoplasmic reticulum membrane of skeletal muscle fibers. It facilitates excitation-contraction coupling, a process where depolarization triggers the release of calcium, leading to muscle contraction.
MHS is inherited in an autosomal-dominant manner, and a single pathogenic variant in RYR1 can be diagnostic for the trait when present in a heterozygous genotype.
Gene #2: CACNA1S and Malignant Hyperthermia Susceptibility
The second locus for MHS is the CACNA1S (calcium voltage-gated channel subunit alpha1 S) gene encoding the α1S subunit of the dihydropyridine receptor, located in the sarcolemma. This functions as the voltage-sensor that is mechanically coupled to and activates RYR1 channels when the sarcolemma is depolarized.
In contrast to many pharmacogenetic tests, there are no star alleles nor diplotypes to be considered for MHS testing. Malignant hyperthermia susceptibility (MHS) is inherited in an autosomal dominant pattern, with rare pathogenic variants, usually missense substitutions, found in either the RYR1 or CACNA1s genes. The determination of whether a variant is pathogenic or benign is complex and relies on various evidence.
Recommendations for Using Genetic Information to Predict MHS Risk

Getting surprised by an MH reaction because you just don’t know until you know? Graaaaand. (Image)
If a person is concerned they may be at risk due to family history, it’s recommended that genetic testing be conducted. If one of the 50 listed variants is found, it’s straightforward to interpret. Anesthesiologists can then make the determination of which drugs to avoid during a procedure and which alternatives are more suited to avoid an episode of MH. However, a negative result or detection of a variant not among the 50 listed requires careful interpretation due to the heterogeneity of MHS.
And then there’s really no way of knowing until the MH reaction occurs. Lovely, right?
Want to see the list of variants? It’s impressive…

Yikes, that’s a lot of possible gene combinations. But I wanted you to be able to see what these variants look like! (Image)
Hopefully you feel a little more informed about this relatively rare (but not rare enough) surgical complication. Plus, bonus, you hopefully know more about inhaled anesthetics than you did before!
The tl;dr of Pharmacogenomics and Precision Medicine
Wow. We’ve covered a LOT of info. And in true infomercial form, BUT WAIT! THERE’S MORE!! There will be a third and final installment of this series coming your way very soon.
To summarize some major takeaways from Part 2 here…
In addition to other individual patient characteristics and non-clinical factors, warfarin dosing is influenced by genetic variations in CYP2C9, VKORC1, CYP4F2, and the CYP SNP CYP2C rs12777823. (Ha, say that fast 3 times in a row…CYP SNP…”sip snip”…)
Risk of statin-associated musculoskeletal symptoms (SAMS) is partially influenced by genetic variations in SLCO1B1, ABCG2, and CYP2C9.
Malignant hyperthermia susceptibility appears to be dependent on the presence of variations in the RYR1 gene, as well as potentially in the CACNA1S gene. The syndrome is triggered upon exposure to inhaled anesthetics or succinylcholine.
Management algorithms that incorporate this genetic information exist for both warfarin and statins, but clinical judgment and considering genetics as just one piece of the puzzle are essential practices.
There you have it! Stay tuned for Part 3, which will include blockbusters like chemotherapy, opioids, and abacavir!!