
Complications of Cirrhosis: Part 1
Steph’s Note: Everybody always wants to talk about the kidney beans. (Which, don’t get me wrong, are HUGELY important in the world of drugs.) But why does nephrology get an entire dedicated therapy module in pharmacy school, whereas that other necessary pharmaceutically-relevant organ might get one or maybe 2 lectures?
Which one is that? THE LIVER!
For being the metabolic powerhouse of the body, we sure don’t spend much time learning about it. So it’s time to take a couple posts to get everyone up to speed on what happens when the liver’s not up to snuff. But first, because y’all probably know me well enough by now to know we HAVE to talk structure-function, we’re going to do a little pathophysiology to start Part 1 of this series.
We combined ALL of the posts in this Liver series AND our Kidney series into a single PDF. If you’d like a downloadable (and printer-friendly) version of this article, you can get one here
Why Should We Care about the Liver?
As one of my preceptors taught me during my PY4 rotations, it’s like this - the liver has 4 main functions in the body:
Synthesis of many proteins, including albumin and clotting factors
Glucose regulation (gluconeogenesis and glycogen storage)
Production of bile and metabolism of nutrients
Detoxification of blood
Of course, this is a simplification of the real deal. In reality, over 500 vital processes have been attributed to the liver. But for the sake of not going cross eyed, we’ll just dump them into my preceptor’s 4 buckets to get an idea of just how essential the liver is.
A Quick Review of Liver Anatomy
The liver is in the RUQ (right upper quadrant) of the abdomen. It is made up of 2 lobes, each of which has a whole bunch of hexagonal “lobules”.
Each lobule consists of hepatocytes (cells) and a circulatory system that allows for material exchange (UBER important to pharmacists because “materials” = DRUGS). This circulatory system includes blood flow into the lobule sinusoids via the hepatic artery (provides oxygen to the hepatocytes) and via the portal vein (brings all the nutrient goodies from the GI system).
Material exchange happens in the sinusoids, which are designed for this purpose with hole-y membranes. Then the sinusoids dump whatever’s left into the central vein, which then dumps into one of the 3 hepatic veins, all of which dump into the inferior vena cava, which goes back to the right heart. Some leftovers leave the liver via the bile ducts instead of blood flow. The bile ducts excrete metabolic by-products (like drug metabolites) into the GI tract to be eliminated in the feces.
For visual peeps:
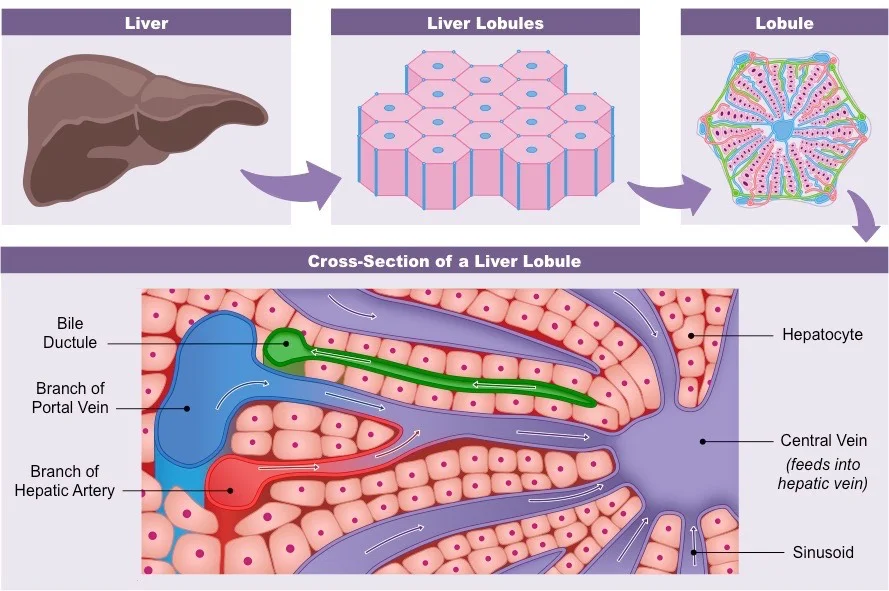
Liver structure, ooo la la lobules! (Image)
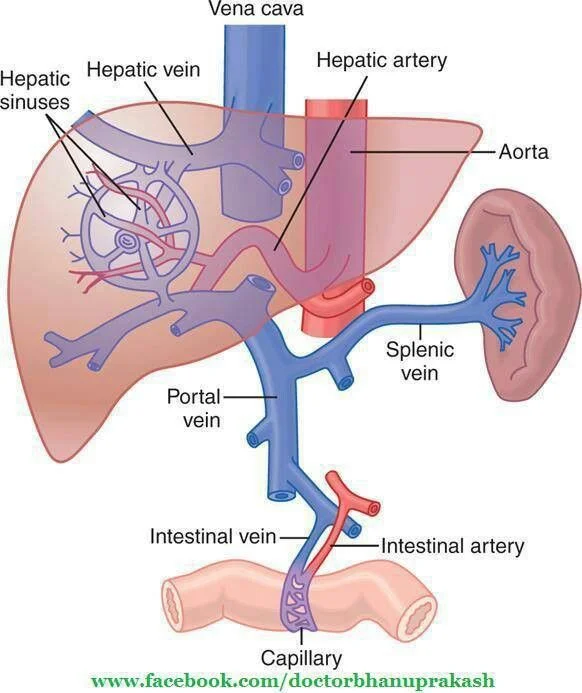
Hepatic blood flow. Note how there are 2 sources of blood going TO the liver: the hepatic artery bringing oxygenated blood to the hepatocytes and the portal vein bringing nutrient (or drug)-rich blood from the GI tract. (Image)
Ok, now that we have the anatomy down pat, let’s talk about what happens when things go wrong.
What is Cirrhosis?
Cirrhosis results when chronic insults to the liver finally cause irreversible fibrosis. What’s fibrosis? Basically, scar tissue. Scar tissue from the liver healing from all those insults.
Depending on the degree of scar tissue (or fibrosis) in the liver, all of the above beautiful anatomy can become distorted. These changes in the cellular architecture lead to inability of the liver to perform its duties AND can affect blood flow within the liver. Hold onto that for a hot sec.
It’s important to think of this fibrosis on a spectrum rather than an all or nothing approach. Patients may have a mild degree of fibrosis, which contrary to previous understanding, can actually be reversible. It’s not until the injury is prolonged and/or progressive that the fibrosis becomes less reversible/irreversible and is deemed cirrhosis.
What kinds of insults or risk factors are we talking?
Mostly things like chronic hepatitis B or C infections, chronic alcohol use, or non-alcoholic steatohepatitis or fatty liver disease (aka NASH or NAFLD). There are others, like autoimmune hepatitis, but the 3 above are the major implicated factors in development of cirrhosis. Note how these are potentially modifiable risk factors, meaning we can try to do something about them. We now have successful (albeit pricey) treatments for hepatitis C infections. Alcohol use disorder can be improved using medications and behavioral therapies. Treatments for NASH center around weight loss and associated medications.
Prognosis is very different for people on different points of this fibrosis spectrum, so educating patients in the early stages about reversible risk factors (and treating those risk factors that we can) is essential.

The spectrum of liver fibrosis. Note how early fibrosis can be resolved, whereas more severe fibrosis/cirrhosis can only be managed. (Image Credit: Semantic Scholar)
So how many people are we talking, and why do we care?

Triple mortality, you say???
As of 2017, cirrhosis was the 11th leading cause of death in the United States according to the CDC. (Sure, those popular kidney beans rank two above at 9th, but the liver deserves attention too!).
And even though cirrhosis due to viral infections is expected to decline given the advent of targeted treatments, the rise in liver disease due to NASH and alcoholism is still worrisome, with a predicted TRIPLING of cirrhosis deaths by 2030.
Yowsers. THAT’S why we need to chat. And keep chatting. Viva la liver!!
For more details, the American Association for the Study of Liver Diseases (AASLD) is our resource for treatment guidelines. Alternatively, the European Association for the Study of the Liver (EASL) released a nice, comprehensive management document in 2018, although (caveat) some medications mentioned are not available in the US. Regardless, between the two organizations, one can get a decent gist of treatments and future directions. This 2016 NEJM article also has a good update on some of the primary literature that was released after several of the AASLD guidelines.
But we at tl;dr are here to save you some time reading (or at least give you a baseline so then you can dig into the details!).
Portal Hypertension: The Source of (Almost) All Evil
In order to move on to the ways that cirrhosis can decompensate, you gotta understand the changes in blood flow that occur. (Remember how we mentioned above to hold onto those cellular architecture implications a hot second ago? Well this is why.)
When fibrosis results in those architectural changes in the sinusoids of the lobules and the lobule metabolic functions fail, there’s basically a road block against blood flow through the liver. This intrahepatic resistance leads to increased pressures in the portal vein, aka portal hypertension.
This is then read by other areas of the vasculature, mainly the splanchnic arterial system, as a need to dilate to alleviate the backup pressures. So via release of nitric oxide and other substances, portal hypertension leads to splanchnic vasodilation.

The vicious cycle of portal hypertension (Image)
The reduced pressures caused by splanchnic vasodilation are also subsequently read by other organ systems as hypotension and a need to increase effective circulating volume. The baroreceptors in the kidneys freak out about this perceived lack of volume, and they activate the renin-angiotensin-aldosterone system (RAAS) by release of renin from the juxtaglomerular complex. This leads to a several main buckets of effects:
arteriolar vasoconstriction via sympathetic activation by angiotensin II
sodium (and therefore water) retention via
angiotensin II action on Na-H channels in the proximal convoluted tubules
release of aldosterone from the adrenal cortex (after stimulation by angiotensin II)
water retention via release of antidiuretic hormone (ADH) from the pituitary gland (also stimulated by angiotensin II)
The resultant hypervolemia actually leads to increased splanchnic blood flow, which (surprise surprise) only leads to further/worsening portal hypertension.

Just like the hateful coagulation cascade, you thought you could learn and forget the RAAS after the exam… Nope! (Image)
Portal hypertension in and of itself can lead to several disease complications, including varices formation and bleeding (gastric or esophageal), hepatic encephalopathy, and ascites, which in turn can lead to spontaneous bacterial peritonitis (SBP).
The renal vasoconstriction caused by this regulatory freakout can lead to another complication of cirrhosis: hepatorenal syndrome (HRS).
Other complications of cirrhosis can be related to some of its synthetic and metabolic functions. For example, coagulopathies may develop given the liver’s role in making clotting factors. The decreased metabolic function also plays a role in the aforementioned hepatic encephalopathy.
Let’s go ahead and forge on to ascites and SBP, but then we’ll save the other complications for Part 2 of this series. But make sure you have a good grasp on portal HTN since it (and its friend angiotensin II) are major players.

One could almost argue that angiotensin II is the source of all evil… But don’t be hatin’. It’s just trying to do its job.
Cirrhosis Complication #1: Ascites
We’ve already established that portal hypertension leads to hypervolemia and increased splanchnic blood flow. What happens with this extra fluid and increased hydrostatic pressure?
Things get leaky.
Increased production of lymph fluid in the liver and splanchnic organs leads to leakage from organs and mesenteric vessels into the peritoneal space, aka the abdominal cavity. This fluid accumulates and voila, ascites.
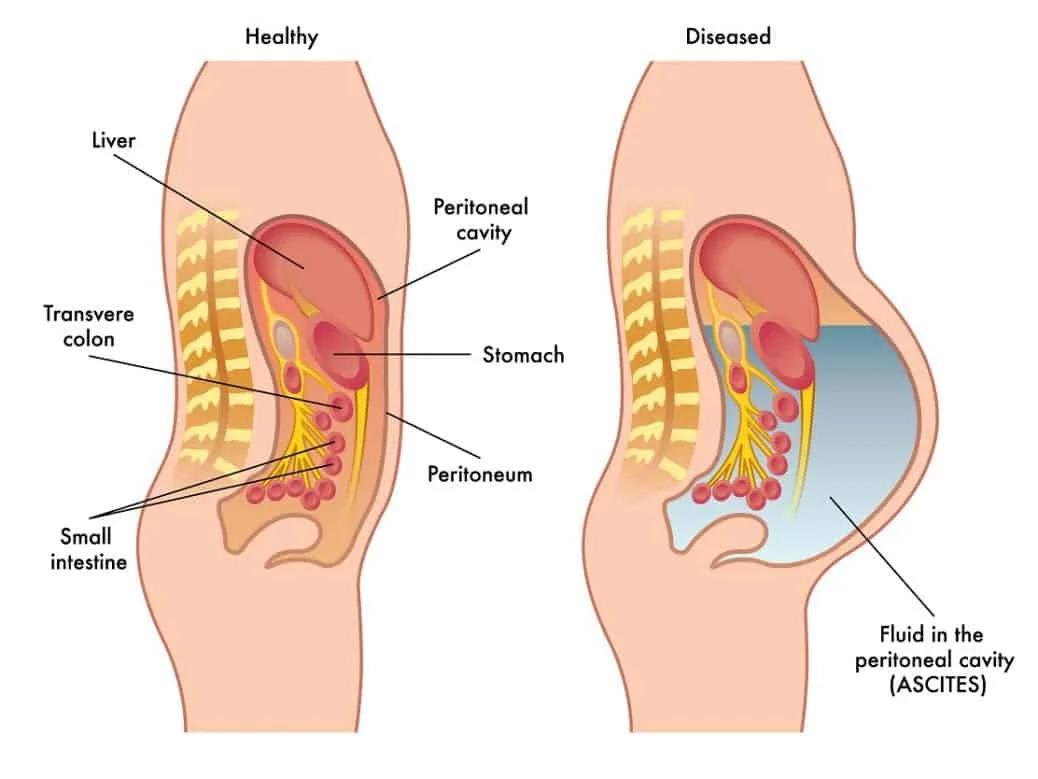
Notice how the fluid is accumulating around the organs and blood vessels in the peritoneal space. (Image)
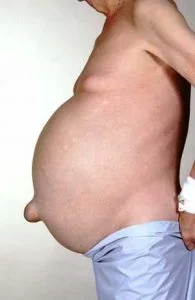
A real look at ascites. (Image)
Now, just btw, there are other causes of ascites besides liver disease (although liver accounts for the majority at ~85%). But some of the other causes may include cancer (including lymphoma and/or those with intra-abdominal metastases), bowel perforation, congestive heart failure with pulmonary hypertension, and pancreatitis, to name a few.
So how do we know if portal hypertension and liver disease are the sources of a patient’s ascites?
We tap that. Mmmhmm.
That’s right. A needle is inserted into the peritoneal space, and then fluid is removed using a syringe. Or (if the volume is large enough) the fluid drains into collection containers. This procedure is called a paracentesis.
Volume removed varies greatly! Sometimes 500 mL is all that’s removed by paracentesis, but some patients can have on the order of 8-9 LITERS (or more) removed!
That’s a LOT of fluid to carry around.
Once this fluid is collected, it can be sent for several different tests to aid with diagnosis. One of these is a measure of the amount of albumin in the ascitic fluid. This number is compared against the serum albumin value, and the result is the SAAG, or Serum Ascites Albumin Gradient.
A SAAG >1.1 is predictive of portal hypertension with 97% accuracy. Not too shabby!
What this ratio does NOT do is tell you anything about the cause of that portal hypertension. So buyer beware, interpret cautiously.
The ascitic fluid can also be qualitatively assessed. Is it clear or cloudy? What color is it?
Quantitative assessments can determine number of white blood cells, specifically neutrophils (or PMNs, polymorphonuclear leukocytes). If the PMN count is >250 cells/mm3, that may be indicative of infection and warrants at least empiric initiation of antibiotics. (More in a few).
The final pharmaceutically relevant test that can be done on ascitic fluid is a culture. Find out what (if anything) is growing in that belly.
Before we go too far down the road of possible infection (although I promise we will!), we need to chat about how ascites is medically managed outside of a paracentesis procedure. While there are patients who require serial, periodic paracenteses in order to remain asymptomatic, many people can be managed with medications.
So we have a fluid overload situation… what kinds of medications would you expect to use?
Ding ding ding, that’s right! Diuretics!
Pharmacologic Management of Ascites

The anatomy of a nephron. Pay special attention to the fact that the Loop of Henle comes before the distal convoluted tubule in terms of urine flow. (Image)
Oops, one quick thing about non-pharmacologic management: sodium restriction. Patients with liver disease who are prone to ascites accumulation should strive to consume no more than 2g of sodium per day in order to lessen fluid retention. More restrictive regimens often lead to malnutrition (because who can find palatable food without at least some salt!?!?).
Anyways. Back to meds. There are two different types of diuretics used in the management of ascites: aldosterone receptor antagonists (aka potassium sparing diuretics) and loop diuretics. Y’all know I love me some mechanisms, so let’s start there.
Aldosterone receptor antagonists (ARAs) do just that - they block the effects of aldosterone in the distal convoluted tubule of the nephron, thereby decreasing Na (and hence water) reabsorption and also impairing potassium excretion. For patients with cirrhosis, the main ARA is spironolactone.
Loop diuretics (furosemide, torsemide, and bumetanide) work on the Na-K-Cl ATPase in the thick ascending Loop of Henle to block Na reabsorption and cause Na and water excretion. Collateral effects include loss of potassium, chloride, calcium, and even magnesium.
However, because Na that is blocked from being reabsorbed in the Loop of Henle can still be reabsorbed in the downstream distal convoluted tubule AND because we know aldosterone is upregulated by the kidney’s RAAS freakout, the ARAs are actually thought to be the more important diuretic class of the 2 in patients with ascites due to portal hypertension. The loop diuretics are really there to balance out potassium levels that can be elevated by the ARAs.
That potassium sweet spot is usually achieved by giving starting diuretic doses of spironolactone 100mg daily and furosemide 40mg daily, both PO. These doses can be titrated up usually about every 3 days to a max of 400mg and 160mg per day, respectively. BUT you should definitely monitor potassium levels and adjust this ratio as needed to correct hyper- or hypokalemia.
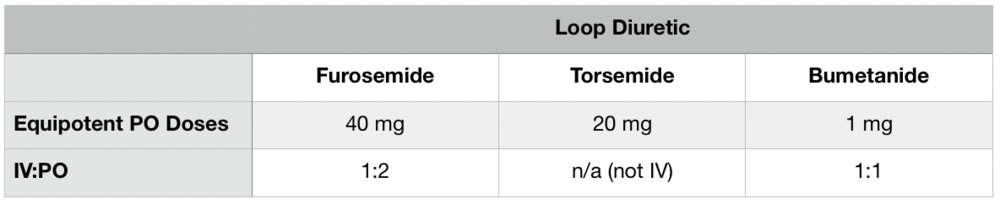
Also, pro tip, be sure to account for differences in furosemide bioavailability in settings of fluid overload. It may be beneficial to use an alternative loop diuretic with better bioavailability (torsemide or bumetanide)!
When patients aren’t sufficiently responsive to diuretics and/or they have accumulated a significant volume, it may be necessary to do therapeutic paracenteses (as opposed to the above discussed diagnostic paracentesis). Same procedure though. The only additional note about paracentesis to make here is this: if volumes greater than 5L are removed (aka a Large Volume Paracentesis or LVP), it’s standard procedure to give albumin afterwards. The general rule of thumb is 6-8 g of albumin per liter of fluid removed.
Why?
You can imagine what happens to the status quo equilibrium when a boatload of volume is removed during paracentesis. The body’s like, whoa, I had this figured out, and you just took one whole side of the fluid equation away! So then whatever’s left in the intravascular space then attempts to equilibrate with the peritoneal space. And BAM. Hypotension due to intravascular depletion. And then the kidneys get angry.
So what do we do to mitigate this?

(Image)
Albumin to the rescue! Albumin is a negatively charged protein. It’s big, it’s stuck in the vasculature because it doesn’t really fit all that well through the exit doorways. So when infused into the vasculature, it stays there and attracts positively charged sodium molecules. And what have we established… where sodium goes, water follows. So albumin is thought to help keep fluid in the intravascular space, especially during the post LVP period. That is, it helps sustain/raise the colloid osmotic pressure (COP), or oncotic pressure.
A final note about ascites before we move on. Regardless of how well a patient’s kidney are doing, NSAIDs aren’t really a good choice for analgesia. They not only cause sodium retention, which is counterproductive to EVERYTHING we just talked about, but they decrease the synthesis of prostaglandins that are essential to renal vasodilation (of the afferent arteriole). So NSAIDs = bad for patients with cirrhosis and ascites.
Cirrhosis Complication #2: Spontaneous Bacterial Peritonitis
Spontaneous bacterial peritonitis (hereafter known as SBP because ain’t nobody got time for that long name) is one complication of ascites. So it’s like 2 degrees of separation from portal hypertension…?
SBP refers to an infection of the ascitic fluid. It’s not entirely clear how this happens, but one of the theories is related to bacterial translocation from the gut with all the leaky and altered vasculature. Other contributory theories include dysfunction of the RES (reticuloendothelial system), decreased phagocytosis, and diversion of inoculated/colonized blood to abnormal locations due to shunts.
Essentially, the immune system is defective in patients with cirrhosis, and physiology seems to allow bacteria to be places they shouldn’t.
Of note, SBP is spontaneous. This is in contrast to secondary peritonitis, in which there’s some inciting factor for the inoculation of the peritoneal fluid. Examples of secondary peritonitis include infections arising after abdominal surgery with perforations or abscesses. It’s crucial to differentiate these because secondary peritonitis requires surgical intervention (no ifs, ans, or buts).
Main point: in secondary peritonitis, somebody opened something up and either introduced bugs or allowed them to migrate. There’s no obvious spider bite that creates Spiderman in SBP.
Although the in-hospital mortality rate has drastically improved in recent years (one study estimating about 18%), especially with early recognition and treatment, one and 2 year mortality rates are still quite high (70% and 80% respectively) after an episode of SBP. Serious patient impact.
Clinical Presentation and Diagnosis of SBP
SBP can be asymptomatic (and even culture negative, just to make things even trickier). Or it can look like full blown sepsis. Things tend to go better if treatments are started earlier rather than later. Surprise. So how do we know whether or not to start treatment?
Ok, remember how a few minutes ago we talked about some of the quantitative and qualitative assessments that ascitic fluid could undergo after paracentesis? Like cell counts and cultures?
These are our clues!
SBP is diagnosed when the ascitic fluid absolute neutrophil count is >250 cells/mm3. (Most of the literature calls these PMNs). Cultures of the ascitic fluid may or may not be positive. Blood cultures are positive in only about a third of patients with SBP. (And you certainly won’t know anything about these right off the bat since cultures have to cook). And given the mortality rates we just talked about, do YOU want to risk missing an episode of SBP?
Didn’t think so.
Pharmacologic Management of SBP
Treatment of SBP is 2 pronged (pharmaceutically speaking). First, antibiotics. Second, albumin. We’ll tackle antibiotics first.

Basically, enteric Gram negatives rods, Strep species, and maaaaaybe some Staph. But not super likely on the Staph. (Image)
In order to figure out what antibiotics to use for empiric treatment of SBP (while those cultures are cooking), we need to know a little more about what kinds of bugs we’re targeting. SBP tends to be monomicrobial (which is another distinction from secondary peritonitis, which is often polymicrobial). It doesn’t usually have obligate anaerobes (e.g., Bacteroides) as a causative organism because there’s too much oxygen in ascitic fluid for them to thrive. What the ascitic fluid DOES often grow are the organisms listed to the right.
Best empiric coverage for these non-Pseudomonas Gram negative rods plus Streptococcus?
Ceftriaxone. Buh BAM. One drug covers about 80% of the organisms in this chart. Sure, you miss the MRSA, the Enterococci, any potential Pseudomonas, resistant ESBLs, and the anaerobes, but short of having some culture/antibiotic history on a person, ceftriaxone does a pretty darn good empiric job.
Dosing of ceftriaxone for SBP is slightly controversial with most studies using 2g/day but most clinicians using just 1g/day for this indication. This 2014 study examined exactly this question, finding the differences in one year survival and length of ICU stay to be not significantly different between the doses — when severity of cirrhosis was adjusted for via the MELD (Model for End Stage Liver Disease) score. But can true conclusions be drawn from this retrospective cohort study? Mmmm…

For patients who are cephalosporin-allergic, quinolones are an acceptable alternative. If you read about SBP in older articles, they are forever mentioning this drug called norfloxacin. Then you walk into your topic discussion with your preceptor and recommend norfloxacin for SBP treatment. (Don’t worry, most of us have done it at some point or another. But FYI, norfloxacin is not available in the US.)
So ciprofloxacin is an acceptable alternative; however, it’s important to consider previous patient quinolone exposures, culture history, and individual patient information (renal function, drug interactions, QTc, etc) when reaching for this med.
Now you’ve decided which antibiotic to use, so the next question is how long to treat… SBP treatment duration is a minimum of 5 days. Of course, see how your patient is responding, etc., but this classic 1991 study and this 1998 study both showed that shorter courses seem to be sufficient.
Alright, on to the second prong of SBP treatment: albumin. We already touched on albumin above in the context of intravascular oncotic pressure following large volume paracentesis. Well, it’s basically there to do the same thing in the treatment of SBP - that is, keep enough fluid in the vasculature to perfuse organs, namely the kidney beans.
See, told you so. The kidneys are forever stealing the liver’s limelight.
Much of the mortality associated with SBP is thought to be related to concomitant or associated renal dysfunction. So the idea is that if we can keep the kidneys happy and perfused, perhaps mortality due to SBP won’t be quite so high.
This 1999 landmark study is the source of our current recommendations for use and dosing of albumin in SBP. It used albumin 1.5 g/kg on day 1 of SBP treatment, followed by 1 g/kg on day 3. But, not everyone necessarily needs or gets albumin. The greatest observed benefits in this study were in patients with the following:
Elevated serum bilirubin >4 mg/dL,
BUN >30 mg/dL, and
Serum creatinine >1 mg/dL.
This 2007 observational study seems to reinforce the idea that perhaps not all patients with SBP need to receive albumin therapy.
A quick side note about albumin. Human albumin is available in two different concentrations. There’s 5% (i.e., 5 g albumin in 100 mL of product) and 25% (25 g albumin in 100 mL of product). Which to use in SBP (or after LVP)?
Well let’s think about it. Both concentrations of albumin have ~130-160 mEq/L of sodium. You’ll notice for both SBP and LVP indications, the dosing is in terms of grams of albumin. So in order to achieve a the same dose in grams of albumin, one would have to give a higher volume of the 5% product as compared to the more concentrated 25% product. Therefore, for a particular albumin gram dose, the sodium load will be higher with the 5% product.
Remember all that talk ages ago about dietary sodium restriction and trying to increase sodium excretion?
Well let’s not go and ruin all that by loading patients back up with a bunch of sodium! So 25% is your friend for both post-LVP and SBP indications.
Secondary Prevention of SBP
The problem with SBP (or well, one of the problems…) is that it’s like a bad habit. As much as you keep putting those bags of Tostitos back in the pantry, you know they’re still there. And you’re gonna bust them out again.
Same with SBP. It likes to come back. Once a patient has had an episode of SBP, the recurrence rate in one year has been reported as 69%. Of course, this study was done in the late 80s, and luckily, practice changes (aka implementation of prophylaxis in select groups) have lowered this number significantly. For example, one study showed a one year recurrence rate of about 28% when preventative antibiotics were given.
70% down to 30%. Seems like a no brainer, right?
Welllll, there are a few things to think about even though those numbers are impressive.
Consider the potential collateral damage if you just throw long term prophylactic antibiotics at every SBP patient. The next time they come in sick (because recurrence rates tell us that’s going to happen…), that bug probably isn’t gonna play nicely. It’s going to require bigger gun antibiotics. Perhaps it’s an ESBL Gram negative this time instead of a friendly E. coli. Perhaps it will be an unexpected Gram positive bacteria that we’ve selected for by giving prophylaxis.
So, as always, treatment is not without some measure of cost.
But still. 70% vs 30%. The benefit seems to outweigh the risk in this case, and secondary prevention of SBP is a go. (More in a sec on possible regimens.)
Primary Prevention of SBP
We can’t quite get away from a discussion about SBP without having a quick moment for primary prevention. That is, given it’s mortality and risk of recurrence, should (and do) we try to prevent SBP before a patient ever even has a first episode??
We talked risk/benefit in the secondary prevention section, so now your feelers should be flagging an even harder “mmmm…I just don’t know” vibe, right?!
Good. That means your spidey-sense is on point. Because it’s a difficult scenario!
Studies have been done to try and figure out which SBP patients should receive preventative antibiotics even before their first episode. According to the AASLD 2012 guidelines, primary SBP prophylaxis can be considered in patients with:
ascitic fluid total protein < 1.5 g/dL, AND
serum creatinine at least 1.2 mg/dL, BUN at least 25 mg/dL, or serum sodium at most 130 mEq/L, OR
Child score at least 9 or bilirubin at least 3 mg/dL.
Basically, it’s the low ascitic protein with signs of either impaired kidney or liver function.
The other time when primary prophylaxis is indicated is in the setting of an upper GI bleed in a patient with cirrhosis. Almost 45% of these patients experience an infection, which can impact resolution of the bleed as well as mortality. So, yes, these patients also get antibiotics.

Prophylactic Regimens for SBP
And NOW, we get to the meat. What drugs and regimens to use?
As above when we discussed SBP treatment, most of what you’ll find refers to use of norfloxacin. Even the AASLD, the American Association for the Study of Liver Diseases, references use of norfloxacin. But norfloxacin is not available in the United States (and hasn’t been for years). So that’s confusing!
We just substitute ciprofloxacin though. NBD.
Ciprofloxacin offers a similar range of coverage to norfloxacin - those enteric Gram negatives. Usually, ciprofloxacin 500mg daily is the regimen of choice for prevention of SBP.
Note that daily dosing is preferred, even though previous studies investigated whether higher dose, weekly regimens could also work. But they just seemed to lead to increased resistance. So daily it is.
If your patient has an intolerance to quinolones (or possibly quinolone resistance on the ascitic fluid culture), sulfamethoxazole-trimethoprim 1 double-strength tab daily is an alternative prophylaxis choice for coverage of those enteric Gram negatives.
For the specific case of upper GI bleeds and primary prophylaxis of SBP, in which long term prophylaxis isn’t really necessary (just 7 days!), most of the time we use ceftriaxone since those people are NPO for at least a period of time. But ciprofloxacin would also be a legitimate option if need be based on intolerances.
So that’s prevention of SBP. Basically, choose patients wisely and weigh risk/benefit.
This wraps up Part 1 of Complications of Cirrhosis. Whew, we’ve covered a LOT! Stay tuned for Part 2, when we’ll cover hepatic encephalopathy, variceal bleeds, and even hepatorenal syndrome.
We combined ALL of the posts in this Liver series AND our Kidney series into a single PDF. If you’d like a downloadable (and printer-friendly) version of this article, you can get one here