
An Overview of ACE Inhibitors
Steph’s Note: Welcome to another round of our pharmacology series! This week, we’re tackling ACE inhibitors (ACE-Is). Even though it sometimes seems like the Lisinopril One Man Show in ACE-I world, it’s important to know what other medications belong to this backbone class and when they may prove useful.
According to the CDC’s 2019 report on prescription medication use in adults, about 11% of Americans between 40-59 years old and 21% between 60-79 were on an ACE-I. This class of medications was in the top 3 categories used by middle-aged adults and in the top 5 for 60+ adults.
With a long laundry list of possible indications, including coronary artery disease (CAD) and post-myocardial infarction (MI), congestive heart failure (CHF), diabetes, nephrotic syndrome, chronic kidney disease (CKD), glomerulonephritis, and even migraines, these medications play an integral role in your life as a pharmacist - no matter what setting you’re in.
So what’s the takeaway here?
We absolutely need to know the deal with this class of meds! That’s a LOT of ACE inhibition going on in this world!
So let’s start with the basics…
BTW - We compiled ALL of our Pharmacology 101 posts into one handy, downloadable (and printer-friendly) PDF. You can get your copy of it here.
How Do ACE Inhibitors Work?
To answer this question, we have to bring back that only-slightly-less-hateful diagram of the Renin-Angiotensin-Aldosterone-System (RAAS). (The most hateful diagram award still goes to the coagulation cascade, which - PHEW - we will NOT be breaking out in any way shape or form here!).
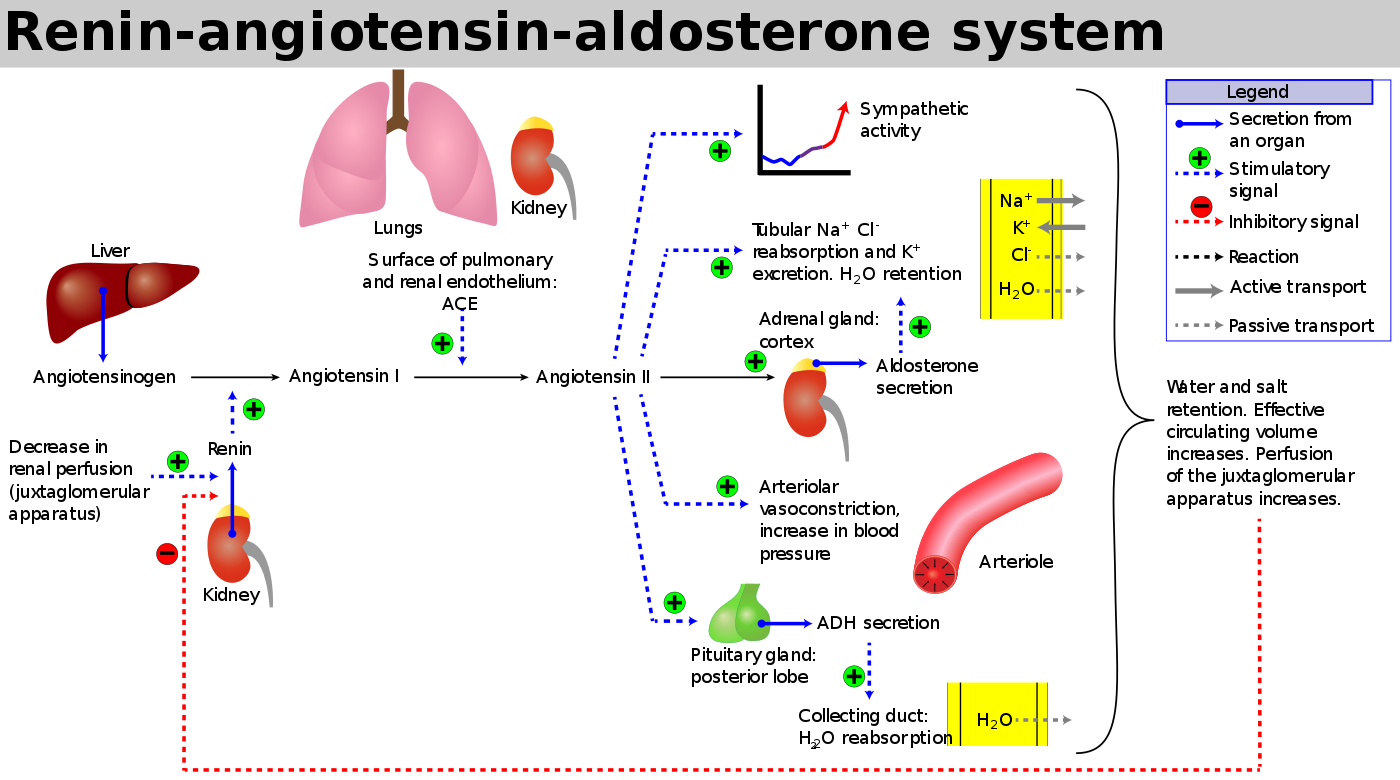
(Image)
We previously busted out this diagram in our Complications of Cirrhosis: Part 1 post, but for today, let’s pick out a few key points from the diagram:
Renin is released from the kidney in response to changes in perfusion pressure. Renin is an enzyme that converts angiotensinogen (a precursor molecule) into angiotensin I.
Angiotensin Converting Enzyme (ACE, aka kininase II) from the lungs and kidneys converts angiotensin I into angiotensin II. This is when all H-E-double hockey sticks breaks loose. Angiotensin II…
Is a potent arterial vasoconstrictor —> increases blood pressure.
Promotes aldosterone secretion from the adrenal gland —> increases sodium and water retention.
Leads to antidiuretic hormone (ADH) secretion from the pituitary —> increases water retention.
Increases sympathetic activity —> increases heart rate and blood pressure.
So even if you can’t always remember all the details from this image, you should at least be able to describe the key players and what converts what (into what). Then, as far as end effects, basically you can’t go wrong with remembering vasoconstriction and sodium and water retention.
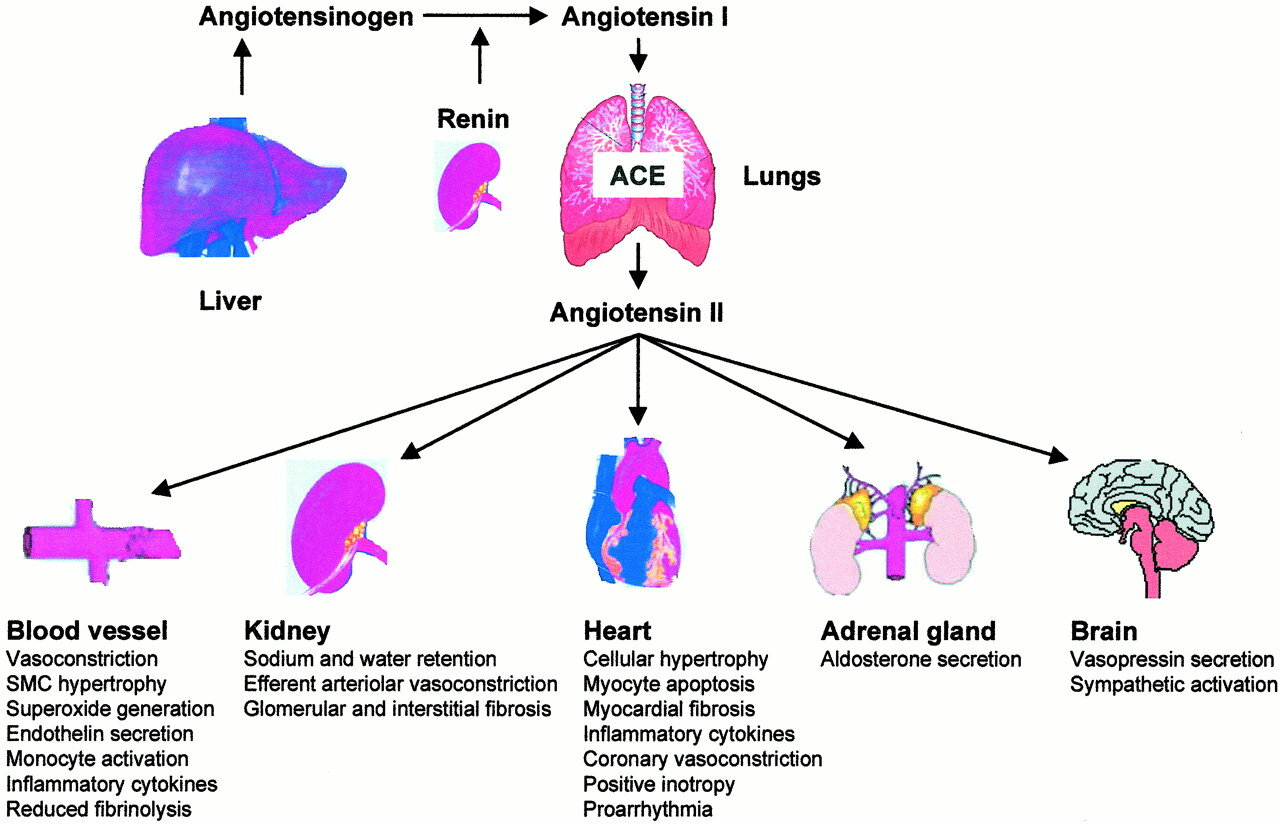
For those visual folks who like a little more detail, here are some of the downstream effects of angiotensin II. (Image)
Disclaimer: We paint RAAS to be this demon of hypertension. But really, it has its physiologic purposes! Don’t forget to thank your RAAS for keeping your organs perfused on those days when maybe you’re a little dehydrated…
Now that we have down pat what RAAS is, we can discuss its inhibition.
Just like their name suggests, ACE-Is block the enzymatic conversion of angiotensin I to angiotensin II, thereby interrupting the cascade of downstream badness and hopefully leading to lower blood pressures, less sodium and water retention, less sympathetic activation, and decreased spillage of protein through the glomerulus into the urine due to high pressures.
Are these medications 100% effective at preventing the downstream effects of angiotensin II and aldosterone?
Unfortunately not. Remember that there are several other pathways for aldosterone release, so even patients who are maintained on an ACE-I may still be experiencing #thebadness. This is called “Aldosterone Escape” and is why combination therapy with aldosterone receptor antagonists is crucial in several disease states, e.g., CHF.
Long story short, with ACE-I, we try to protect the heart. And we try to preserve the kidneys. Pretty important, yay?
Selecting an ACE-Inhibitor
Raise your hand if you’ve ever counted pills for a lisinopril script…
Now everyone high five each other. (Socially distanced, of course.)
Because we all have seen the lisinopril 1 tablet PO daily, #30 or #90 scripts many many times a day. And yes, it’s dirt cheap. But believe it or not, lisinopril does actually have cousins that are used as well for particular reasons, and knowing at least a little bit about each one’s pharmacokinetics just may come in handy. Let’s take a look at the ACE-I family:
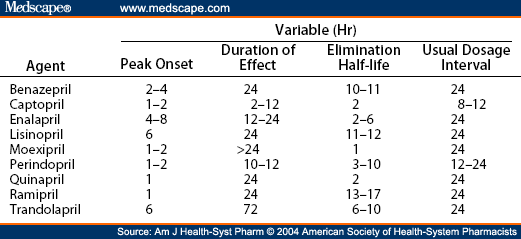
(Image)
In about 10 years of my former inpatient internal medicine life, I ran across each of these at some point or another - EXCEPT moexipril. (That one I honestly just put into LexiComp because I needed to see if it’s even available in the US. FYI, it technically is.)
But maybe friends don’t let friends choose the weirdest name on the list for a recommendation, mmmk?
For the others, can you image scenarios when enalapril’s shorter half life and shorter duration of action may be more desirable than lisinopril? Perhaps it’s the patient who is hypertensive during the day but drops low at nighttime… What about captopril - do you need to be able to titrate meds quickly? Wouldn’t it be nice to make dosing changes within the same day rather than having to wait until tomorrow?
ACE Inhibitor Dose Conversion
Outside of trying to make individualized recommendations for which ACE-I is best, if you work in a hospital, more than likely you will hit The Formulary Wall with these medications at some point. For example, your newly admitted patient is on quinapril at home, but you only have lisinopril and captopril available on formulary. No one can get the patient’s medication from home. And it’s up to you to formulate a plan for substitution during the patient’s admission.
This is undoubtedly one of the hardest things to do as a student or new practitioner. Honestly, sometimes it’s still hard.
If you try to search for “switching between ACE-Is” in Up to Date or Google, you’ll probably just get some charts, most of which look something like the one below. They include some common dosing ranges for each medication and the usual dosing frequencies. But no one straight up tells you exactly how to switch between medications within this class…
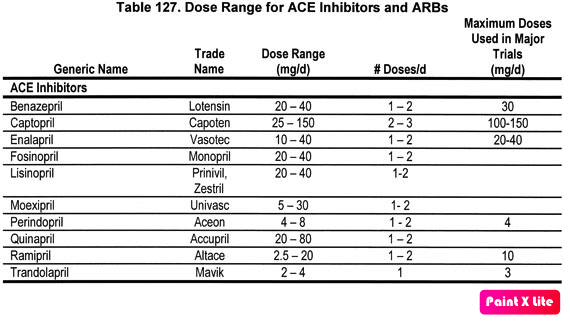
(Image)
Clinical judgment time.
Perhaps start by considering the patient’s home regimen, his current pressures, other pertinent labs, and when the last dose of medication was prior to admission. Was the patient at the low end of the dosing range for that ACE-I? Or was he maxed out? What would “maxed out” look like with your formulary agent, and do you feel comfortable with that in current state? Is he hypertensive now, and how much wiggle room do you have if you overshoot?
You will inevitably have to stake your claim. And then monitor, monitor, monitor to see where you end up. Don’t forget that the initial effects of the medication may not be reflective of the case at steady state (~5 half-lives later).
About that monitoring…let’s chat about adverse effects next.
ACE Inhibitor Side Effects
There are really 4 big adverse effects that we should cover here:
Increased serum creatinine
Hyperkalemia (increased serum potassium)
Dry, hacking cough
Angioedema
Increased Serum Creatinine
Starting with the first, increased serum creatinine is pretty much inevitable when starting a patient on an ACE-I. Think about the mechanism, and then let’s apply that to the kidneys. The decrease in angiotensin II leads to preferential vasodilation of the kidney’s efferent arteriole compared to the afferent arteriole. That means the blood vessel carrying blood away from the glomerulus is more dilated than the blood vessel carrying blood to the glomerulus. What does this do to the pressure within the glomerulus?
Think about water in pipes. Water flowing from a small diameter pipe into a larger diameter pipe is going to be under less pressure in that transition period, right? (As opposed to if water is flowing from a large diameter pipe into a smaller diameter pipe when all that water’s going to get smushed in together in the transition.)
So in the presence of an ACE-I’s efferent arteriole vasodilation, the pressure in the glomerulus decreases. And although this may not be a bad thing on the whole given the overall beneficial effects, it does mean filtration of substances within the glomerulus (from the blood into the nephron) also decreases. So things…like serum creatinine…that are normally filtered out of the blood into the nephron may not be filtered as efficiently, leaving higher levels in the blood.
This is to be expected. In fact, it’s entirely reasonable that a patient’s serum creatinine increases up to 10% when starting on an ACE-I. Even up to 20% over the first couple of weeks may be acceptable.

The combined effects of NSAIDs and ACE-Is on the afferent and efferent arterioles, respectively. WARNING: potential for drug interaction and kidney injury! (Image)
Larger increases than that may indicate that you’ve pushed the dose too hard and dropped the glomerular pressure too far, too fast (rather than letting it acclimate). We would definitely not want to see that 20% increase in serum creatinine after 1-2 doses of an ACE-I - because where will it be when the patient reaches steady state??
ACE-Is used to carry a contraindication in patients with bilateral renal artery stenosis, but this has been downgraded to a warning statement at this point. Controversy exists regarding risk vs benefit in this population due to the above mechanism of efferent arteriole vasodilation and decreased glomerular pressure, especially in the setting of stenosed incoming blood vessels. (It kind of mirrors the scenario to the right with NSAIDs, except the lack of incoming blood flow is due to stenosed vessels rather than medication effect.)
So the takeaway point here - start with lower doses of ACE-Is and uptitrate carefully over time. (Personally, I cap my initial lisinopril dose at 10mg and increase from there.) Also, watch for drug interactions, such as with NSAIDS, and consider ACE-I use carefully if a patient has renal artery stenosis.
Another takeaway point from this section - just because ACE-Is can cause an increase in serum creatinine does not mean that they can’t be used in patients with pre-existing renal dysfunction. In fact, ACE-Is are generally considered ok to start in patients with baseline serum creatinine levels of up to 3 mg/dL. NOTE that we’re talking CHRONIC kidney disease here, not ACUTE kidney injury. An acutely elevated serum creatinine of 3 mg/dL when a person is normally 1 mg/dL is NOT the same as someone who is chronically elevated.
Hyperkalemia
Next, there’s hyperkalemia, or increased serum potassium. Hyperkalemia is usually classified as a serum potassium level >5 mEq/L. This adverse effect of ACE-Is is a result of these medications’ effects on aldosterone activity. Remember that angiotensin II usually stimulates release of aldosterone from the adrenal cortex, which leads to sodium and water retention in the kidney. Well this sodium and water retention occurs in tandem with excretion of potassium via the action of a Na/K ATPase.
So when we block (at least some of) the activity of aldosterone with the use of ACE-Is, there is less excretion of potassium within the renal tubule. And voila, more potassium hangs around.
Hyperkalemia with ACE-Is isn’t usually something that happens in just any patient out of the blue (although it can). Most of the time, patients have some underlying (perhaps as yet undiagnosed) kidney disease, are taking in extra potassium through diet or supplements, or are taking other medications that contribute to potassium retention. So review that medication list when starting an ACE-I and be sure to recommend regular, periodic lab checks, especially in patients at risk!
Cough
Then, we come to the dry, hacking cough that’s associated with ACE-Is. It’s estimated that up to a third of patients started on ACE-Is will encounter this side effect with increased frequencies in female and Black patients. It can also develop at any point in therapy from the initial days to weeks or months after starting. It is not a dose-dependent effect.
Of note, this cough isn’t actually dangerous - unless you take into account loss of medication adherence because it’s just SO DARN ANNOYING. So where does this cough come from?
Not sure if you noticed, but I snuck in a nugget of a clue above… ACE is also known as kininase II because of its involvement in the kinin-kallikrein system. In addition to the whole conversion of angiotensin process, ACE/kininase II is also responsible for degradation of bradykinin. This substance is actually a strong vasodilator, and it’s been theorized that the anti-hypertensive effects of ACE-I may come in large part from keeping more bradykinin in circulation.

The other half to the ACE and ACE-I story: the Kallikrein-Kinin System. (Image)
Downside - it turns out bradykinin is also pro-inflammatory, and when we cause a build up of it with the activity of ACE-Is, the result is often that dry, hacking, annoying cough.
Pro-tip: If your patient is experiencing an ACE-I cough, this is a great time to consider use of another class of RAAS medications - the Angiotensin Receptor Blocker (ARB). These medications exert their therapeutic actions by antagonizing activity of angiotensin II at its receptors, leaving ACE intact. This allows for continued normal kinin-kallikrein processes and bradykinin degradation. Hence, no cough!
Angioedema
And finally, the last of the big 4 ACE-I adverse effects… Angioedema. You may remember this previous angioedema post, and while hereditary angioedema may clinically resemble ACE-I induced angioedema, they have distinct mechanisms.
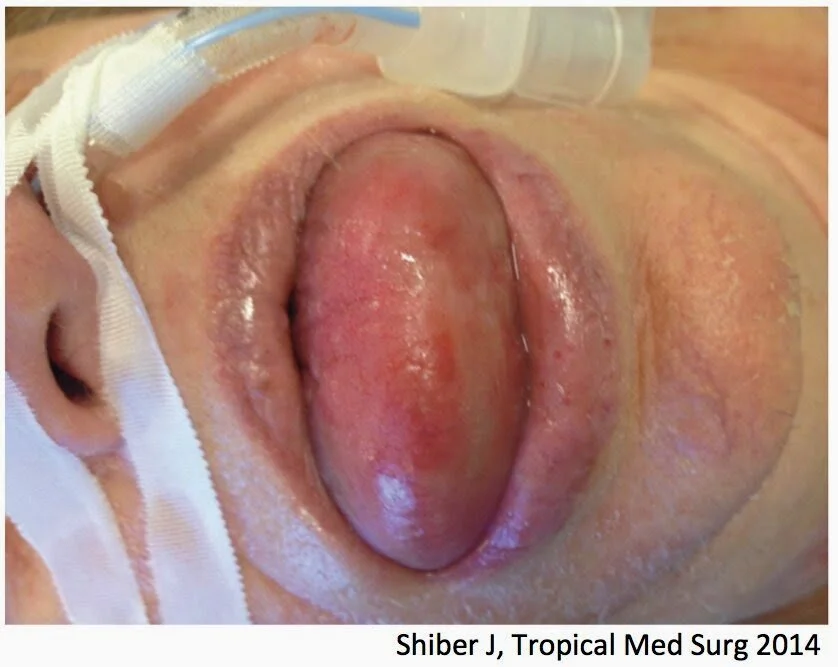
One example of ACE-I-associated angioedema, in which the tongue was largely affected. Alternatively, the lips and cheeks may also swell. (Image)
ACE-I angioedema is bradykinin-mediated, much like the dry, hacking cough of above. To borrow from that previous post, ACE-I-associated angioedema may affect patients at any age and any dose--even if patients were previously asymptomatic and tolerating the ACE-I!
In contrast to the cough, angioedema can absolutely be a life-threatening emergency. Although it’s not incredibly common (one study estimated that 0.7% of patients on ACE-Is developed angioedema over the course of 5 years), it is imperative to counsel patients on early signs and symptoms so they know to seek urgent medical attention.
For example, let me tell you about this time my dad started lisinopril…
I was but a first-year pharmacy student, still in the stages of learning all these good adverse effects and counseling points for medications. My dad had recently experienced a prolonged hospitalization, during which he was started on lisinopril for hypertension.
After a month in the hospital not being able to eat much “normal” food, he was super pumped to get some Bojangles after discharge. (Not that I’m saying we typically eat Bojangles and that’s “normal” fare for our family - refer to Pharmacology 101: Statins. I promise we believe in veggies).

There it is - heaven.
Also, if you don’t know what Bojangles is - let me tell you this. It’s a bit of southern fried chicken heaven, courtesy of my home state of NC. Supreme dinner, fries with extra seasoning (just dump it on, please), BBQ sauce, and iced tea. Which everyone knows means sweet tea.
Anyway, I digress.
Dad finally got his Bojangles, and we were all sitting around the table eating fried heaven. He started talking a little oddly. Like a little muffled. Perhaps like he bit his tongue or the spices got to him. We gave him a little heck and kept munching.
Fast forward 5 minutes. Dad was really starting to sound weird. We looked in his mouth to see if he had any irritations from the food, and interestingly, his tongue looked a little larger than normal. We headed to the car, thinking we needed to take him to the clinic to make sure everything was ok.
Mind you, the clinic was about 20 minutes away.
By the time we reached the health system, clinic was no longer an option. We headed for the ER because his tongue was FILLING HIS MOUTH, and he was having a difficult time breathing.
Luckily, they were able to start management right away and didn’t have to do any invasive procedures to maintain an airway, but I can tell you how important it is for patients to know that this is possible. What if we’d waited just a little longer at home? What if there had been a traffic jam??
So perhaps not the most common of adverse effects, and we don’t want to scare people away from taking their ACE-Is. But I present it much like I would an allergy when counseling - rare, but possible, and people should know what to look for.
Soapbox over, promise. But maybe my story will help this stick in your head for this class of meds!
Final thoughts on ACE-I-associated angioedema are regarding treatments. Many of these drugs are actually FDA-indicated for hereditary angioedema, but studies have been conducted to see if they could have utility in the ACE-I version. These therapies include medications like icatibant and ecallantide. Unfortunately, studies haven’t shown a particularly overwhelming benefit in favor of using these medications for ACE-I-associated angioedema. Hopefully there will be more to come on treatments for this potentially fatal adverse effect, even if it’s not overly common.
Pro tip #2: While switching from an ACE-I to an ARB may solve the annoying cough problem, it may or may not be the solution for a patient who has experienced ACE-I-associated angioedema. It’s still unclear what the true cross-over risk is between these two classes and whether time lapsed from event plays a role. One 2008 meta-analysis estimated that as many as 10% of patients will experience cross-sensitivity reactions from ARBs and ACE-Is, although subsequent studies have since postulated lower percentages.
And there you have it! An overview of ACE-Is. Hopefully this has helped you to understand the why’s behind some of those monitoring and counseling points, as well as how to deal with the lesser known members of the family.
BTW - We compiled ALL of our Pharmacology 101 posts into one handy, downloadable (and printer-friendly) PDF. You can get your copy of it here.